Population-specific polygenic risk scores for people of Han Chinese ancestry
Article Date: 15 October 2025
Article URL: https://www.nature.com/articles/s41586-025-09350-y
Article Image: https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41586-025-09350-y/MediaObjects/41586_2025_9350_Fig1_HTML.png
Summary
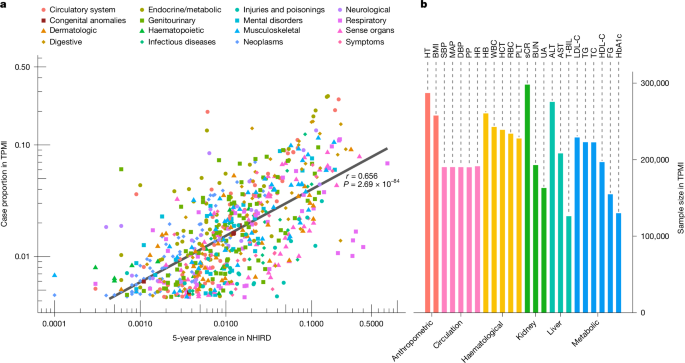
This paper reports large-scale, phenome-wide genetic analyses in people of Han Chinese ancestry using the Taiwan Precision Medicine Initiative (TPMI) cohort. The team genotyped and linked electronic medical records for roughly half a million participants and ran GWAS on 695 phecodes and 24 quantitative traits. They discovered 2,656 independent variant–trait signals (including dozens of novel, population-specific hits), performed fine-mapping and colocalization with expression datasets, identified trait clusters with shared genetics, and developed polygenic risk scores (PRS) optimised for this population.
Population-specific PRS — built and tuned with LDpred2 and combined multi-trait methods — showed strong predictive performance for many traits (for example ankylosing spondylitis AUC ~0.81; type 2 diabetes AUC ~0.64), and TPMI-derived PRS outperformed European-derived models when applied to East Asian samples in external cohorts (Taiwan Biobank, UK Biobank EAS subset, All of Us). Multitrait PRS improved prediction for correlated clusters (cardiometabolic, autoimmune/infectious, kidney-related). The authors discuss limitations (ascertainment bias, incomplete EMRs, underrepresentation of EAS in eQTL resources) and call for broader, diverse datasets.
Key Points
- TPMI assembled genotype and longitudinal EMR data for ~463,447 individuals genetically similar to Han Chinese and analysed 695 phecodes + 24 quantitative traits.
- GWAS and fine-mapping identified 2,656 independent variant–trait associations, including 95 previously unreported associations and many variants rare or absent in European datasets.
- Colocalization and gene-level heritability highlighted pleiotropic genes (eg APOE, ABCG2, KCNQ1) and three main phenotype clusters: cardiometabolic; autoimmune/infectious; kidney-related.
- PRS models (LDpred2 performed best overall) achieved meaningful prediction for many diseases; multitrait PRS (PRSmix+) notably improved cardiometabolic prediction (AUC +0.04 and 1.77-fold r2 increase).
- TPMI-derived PRS validated externally (Taiwan Biobank, UKB EAS, All of Us) and generally outperformed PRS built from UK Biobank European data when applied to East Asian samples.
- PRS explain a measurable share of health-service measures: top PRS models accounted for ~8.5% of variation in clinic visit frequency and ~10.3% of hospitalisation duration between top vs bottom groups.
- Limitations include hospital-based ascertainment bias, incomplete cross-hospital EMRs, limited EAS representation in expression datasets, and insufficient size for some rare subtypes.
Context and Relevance
Modern PRS have been dominated by European-ancestry datasets, limiting transferability. This study shows the value of building population-specific genomic resources: large, local cohorts reveal variants and effect sizes that are invisible in EUR-focused studies and allow PRS that work better for the target population. The TPMI work is particularly important for traits enriched or distinct in Taiwan (for example hepatitis B) and demonstrates that with sufficient sample size and high-quality EMRs, population-specific PRS can approach the predictive power seen in European-based efforts. The findings support a move towards equitable precision medicine by expanding diverse genomic resources and tailoring risk models to ancestry and local disease patterns.
Author style
Punchy: big cohort, clear wins. The paper is a strong demonstration that you can’t just repurpose European PRS and expect optimal results — build the models on the right population and you get better, clinically meaningful prediction. If your work touches genetics, population health or PRS deployment, read the details: variant discovery, fine-mapping, multitrait improvements and external validation are all here.
Why should I read this?
Short answer: because it shows PRS actually work — but only when you build them for the right people. If you want to know how population-specific data changes discovery, fixes missed variants and improves risk scores for East Asian populations (and what that means for real-world screening and prevention), this paper saves you knuckling through a stack of supplementary files. It’s a practical blueprint for making PRS fairer and more useful.