Myocardial reprogramming by HMGN1 underlies heart defects in trisomy 21
Article metadata
Article Date: 22 October 2025
Source URL: https://www.nature.com/articles/s41586-025-09593-9
Image: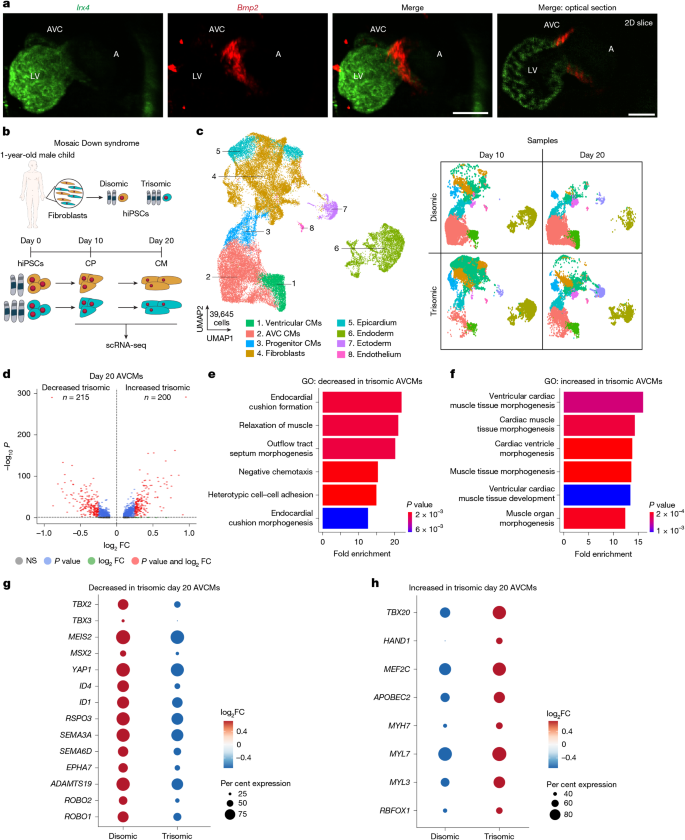
Summary
This study uses isogenic human induced pluripotent stem cells (hiPSCs), single-cell RNA-sequencing and a pooled CRISPR-activation (CRISPRa) CROP-seq screen to identify chromosome 21 genes that, when modestly upregulated, distort cardiac cell identity. The authors find that increased expression of the chromatin-binding protein HMGN1 shifts specialised atrioventricular canal cardiomyocytes (AVCMs) towards a ventricular-like transcriptional state — a change that mirrors trisomy 21 (Down syndrome) cells. They validate HMGN1’s role by allele-specific editing in human trisomic cells and by normalising Hmgn1 dosage in a mouse trisomy model, which rescues AVCM gene expression and reduces the incidence of ventricular septal and outflow tract defects.
Key Points
- Trisomy 21 causes AVC myocardium to lose its specialised identity and acquire ventricular-like gene expression, which likely contributes to septal defects common in Down syndrome.
- A focused CRISPRa CROP-seq screen of 66 candidate chromosome 21 genes identified HMGN1 as a top hit that can recapitulate trisomic AVCM reprogramming when upregulated ~1.4–1.6-fold.
- Allele-specific deletion of one HMGN1 copy in trisomic hiPSCs restores AVCM transcriptional programmes toward the disomic state.
- HMGN1 occupies distal regulatory regions near key cardiac transcription factors (for example TBX2 and TBX20) and motifs for MEF2, GATA, TEAD, CTCF and JUN/FOS, suggesting a chromatin-mediated mechanism.
- In the Dp1Tyb mouse model of Down syndrome, reducing Hmgn1 from three to two copies corrects AVCM gene dysregulation and significantly reduces the incidence of ventricular septal and outflow tract defects.
- The multimodal pipeline (isogenic hiPSCs, single-cell genomics, CRISPRa screening, CUT&RUN and mouse genetics) demonstrates a scalable strategy to map dosage-sensitive genes within large aneuploid regions.
Content summary
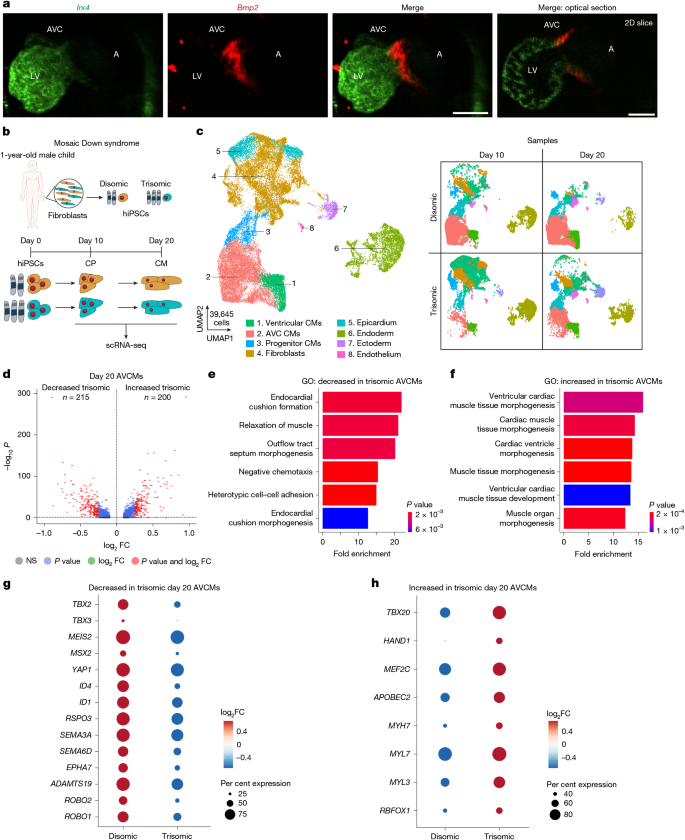
The authors started with isogenic disomic and trisomic hiPSC lines from mosaic individuals to avoid background genetic confounders. Directed cardiomyocyte differentiation followed by scRNA-seq revealed a reproducible shift: trisomic AVCMs downregulated AVC-specific genes (for example TBX2/TBX3 signalling and extracellular matrix components) and upregulated ventricular programmes (TBX20, HAND1, MYH7), consistent with loss of AVC identity.
To functionally screen chromosome 21 genes at physiologically relevant upregulation levels, they engineered disomic hiPSCs with dCas9-VPR and performed a pooled CROP-seq CRISPRa library targeting 66 candidate genes expressed during cardiogenesis. Using a trained statistical classifier that scored cells on a ‘trisomic’ transcriptomic axis, HMGN1 emerged as a top candidate. Arrayed CRISPRa validation showed that HMGN1 activation most closely reproduced the trisomic AVCM-to-ventricular shift.
Complementary allele-specific knockout in trisomic hiPSCs reduced HMGN1 expression to disomic levels and partially restored AVCM gene programmes. CUT&RUN of FLAG-tagged HMGN1 in cardiomyocytes localised HMGN1 to distal regulatory elements enriched for cardiac TF motifs, supporting a chromatin-mediated reprogramming mechanism.
Finally, normalising Hmgn1 dosage in the Dp1Tyb mouse model corrected the AVCM transcriptional signature in vivo and rescued the increased incidence of ventricular septal and outflow tract defects by microCT assessment and reduced neonatal lethality — providing morphogenetic evidence that Hmgn1 dosage contributes to the cardiac phenotype of trisomy 21.
Context and relevance
This work addresses a central challenge in aneuploidy biology: pinpointing which of many dosage-altered genes drive specific developmental defects. Cardiovascular malformations are common and clinically important in Down syndrome; identifying HMGN1 as a dosage-sensitive chromatin regulator links gene-dosage effects to cell-type-specific epigenetic reprogramming of myocardium. The paper also showcases a generalisable pipeline — isogenic hiPSCs, single-cell readouts, CRISPRa perturbations and in vivo genetics — that can be applied to dissect other aneuploidy-associated phenotypes and complex loci where multiple genes collaborate.
For researchers interested in congenital heart disease, epigenetics or Down syndrome, the result suggests new mechanistic hypotheses and potential intervention points: modulating chromatin regulators or their downstream programmes during critical developmental windows might alter defect penetrance. It also reminds us that modest (physiological) expression changes matter — and that CRISPRa is a useful tool to model those changes.
Author style
Punchy: this is a strong, methodically rigorous study that elevates HMGN1 from a chromosome 21 bystander to a bona fide dosage-sensitive factor shaping AVC myocardium and congenital heart disease risk in trisomy 21. The genetic and single-cell evidence — human cells plus mouse rescue — makes the finding hard to ignore.
Why should I read this?
Quick take: if you’ve any interest in Down syndrome, congenital heart defects or how tiny changes in gene dosage rewire cell identity, read this. It's one of those papers that actually links a specific trisomic gene to a clear developmental defect using human cells and a mouse rescue. Saves you the slog of wading through dozens of partial-trisomy studies — the authors do the heavy lifting and give you a concrete target and a plausible mechanism.