A pangenome and pantranscriptome of hexaploid oat
Summary
This Nature paper presents a chromosome-scale pangenome and pantranscriptome for hexaploid oat (Avena sativa). The authors assembled and annotated 33 genomes (the PanOat panel) and generated transcriptome data across six tissues for 23 of those lines. They produced a gene-expression atlas, defined core/shell/cloud gene compartments, mapped gene presence–absence variation (PAV) and surveyed large structural variants (SVs). Key findings include subgenome-biased expression (C-subgenome underexpressed), compensatory upregulation following gene loss (more common between the closely related A and D subgenomes), and multiple major chromosomal rearrangements that impact breeding — notably a large pericentric inversion on chromosome 7D linked to earlier heading and a 2A/2C homeologous exchange associated with widely used Australian semi-dwarf varieties. The datasets and genome browsers are publicly available (GrainGenes, ENA projects), providing immediate resources for oat research and genomics-assisted breeding.
Key Points
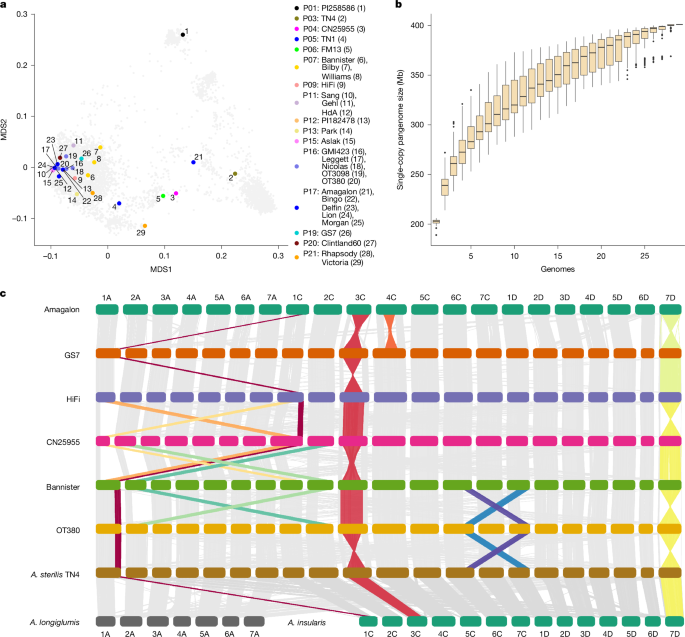
- Thirty-three chromosome-scale oat genomes were assembled and annotated; transcriptomes from six tissues were profiled for 23 lines to build a pantranscriptome.
- The pangenome partitions genes into core (present in all), shell (in many) and cloud (unique) categories; core genes are enriched for essential processes while shell/cloud contain defence and adaptive functions.
- Subgenome expression bias: C-subgenome genes show lower mean expression than A or D, and A–D homeologues compensate more readily for each other after gene loss than C does.
- Large structural variants are common and functionally important: a ~420–450 Mb pericentric inversion on 7D is associated with earlier flowering, and a 2A/2C homeologous exchange (likely linked to historical mutation breeding) is frequent in successful Australian varieties and correlates with yield advantage in trial data.
- Synthetic-derived and breeding lines carry homeologous exchanges and chromosome substitutions (for example 6D replaced by 6A), which substantially shift expression patterns and may be harnessed or avoided in breeding.
- SVs suppress recombination and cause segregation distortion in crosses; the study provides maps and SNP/k-mer markers to help breeders choose parents and interpret mapping results.
- All data, genome browsers (33 accessions) and tools (BLAST, eFP expression pages) are deposited in public repositories (ENA, GrainGenes) to accelerate oat improvement.
Context and relevance
Oat is a globally important cereal with rising demand (oat milk and health markets). Genomic resources for oat have lagged behind other cereals because of its large, repeat-rich allohexaploid genome. This pangenome turns that challenge into an opportunity: by delivering multiple reference-quality assemblies plus a pantranscriptome, the work exposes the structural and expression diversity that shapes agronomic traits and breeding outcomes. The study is directly relevant to plant geneticists and breeders working on oat or related polyploid cereals, and to anyone interested in how structural variation and subgenome interactions influence trait evolution and crop improvement.
Why should I read this?
Short version: if you care about making better oats (or understanding tricky polyploid genomes), this paper saves you the legwork. It bundles 33 genome assemblies, a multi-tissue expression atlas, and clear examples of how big chromosome changes affect flowering, yield and breeding. Practical takeaway: maps and markers are now available to diagnose SVs that break crosses or preserve useful allele blocks — handy for breeders and genomicists.