Assessing phylogenetic confidence at pandemic scales
Summary
This paper introduces SPRTA (subtree pruning and regrafting-based tree assessment), a new branch-support measure designed for pandemic-scale phylogenetics. Unlike traditional topological support measures (for example, Felsenstein’s bootstrap or aBayes), SPRTA focuses on the mutational or placement interpretation of branches — i.e. the probability that a descendant subtree truly evolved from a specific ancestor — and is built to scale to millions of genomes. Implemented in MAPLE (and available in CMAPLE/IQ-TREE integrations), SPRTA reuses likelihood calculations already done during tree search and evaluates many SPR placements efficiently. Benchmarks on simulated SARS-CoV-2-like data show SPRTA is both far faster (orders of magnitude less CPU/memory) and better at discriminating correctly inferred mutation events versus erroneous ones. Applied to a 2,072,111-genome SARS-CoV-2 tree, SPRTA highlights large numbers of uncertain sample placements and mutation events, shows how uncertainty affects site-specific mutation counts and lineage origins (Pango assignments), and provides an annotated tree and data on Zenodo for exploration.
Key Points
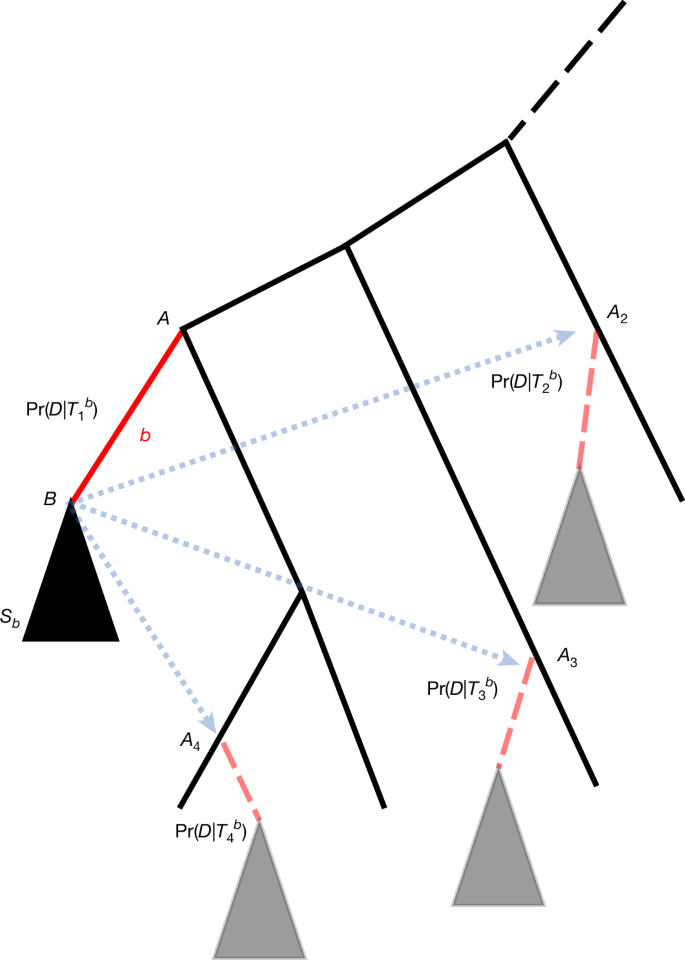
- SPRTA assesses branch support with a mutational/placement focus: the probability that a subtree evolved from a given ancestral node rather than the standard clade-existence interpretation.
- It uses SPR moves (many more alternatives than NNI) and efficient likelihood approximations (as in MAPLE) to remain computationally tractable at pandemic scale.
- SPRTA is robust to rogue taxa (incomplete or ambiguous sequences) and can score terminal branch placements — something topological methods cannot do.
- Benchmarks show SPRTA reduces runtime and memory by at least two orders of magnitude versus popular approaches and better separates correctly inferred mutations from incorrect ones.
- Applied to >2 million SARS-CoV-2 genomes, SPRTA finds substantial uncertainty: many sample placements and inferred mutations have low support, and some Pango lineage origins are plausibly different from the ML tree.
Content summary
Phylogenetic analyses at pandemic scale (millions of genomes) require fast, reliable measures of confidence. Traditional bootstrap and local branch-support measures are either too conservative for low-divergence epidemiological data or too computationally heavy to run on such large datasets. SPRTA reframes branch support as the probability of a mutational origin (placement) and evaluates many plausible alternative placements created by SPR moves. The method filters candidate placements with a cheap fixed-branch-length pass, then refines likelihoods by optimising a few local branch lengths — keeping computations local and fast.
In simulations tailored to SARS-CoV-2, SPRTA consistently assigns very high support to correctly inferred mutation events and lower support to wrongly inferred ones, outperforming other methods for the mutational task. On a real dataset of 2,072,111 genomes, SPRTA ran in hours (post hoc) or can run essentially concurrently with MAPLE tree inference, revealing hundreds of thousands of mutations and many internal and terminal placements with low support. The authors show concrete examples (for example, uncertainty in AY.4 and BA.2.13 origins) where SPRTA identifies plausible alternative histories and quantifies their probabilities.
Context and relevance
For anyone working in genomic epidemiology, viral phylodynamics or large-scale evolutionary analysis, SPRTA provides a practical way to quantify uncertainty that is both meaningful for lineage/mutation interpretation and feasible at pandemic scale. It helps avoid overconfidence in downstream analyses (mutation-rate inference, lineage definitions, transmission reconstructions) by marking which mutations and placements are well supported and which are not. The annotated 2M SARS-CoV-2 tree and code (MAPLE/CMAPLE integrations) are available for inspection and use.
Author style
Punchy: This is a practical breakthrough for pandemic-scale phylogenetics. The authors don’t just propose a statistical tweak — they deliver a tool that scales, is implemented in production software, and exposes real uncertainty in huge SARS-CoV-2 datasets. If you care about reliable lineage or mutation calls at scale, read the details.
Why should I read this
Quick and casual: if you handle pathogen genomes, this paper saves you from false certainty. SPRTA tells you which branches and sample placements you can trust and which you should treat with scepticism — and it does it fast enough for millions of genomes. It’s like a reality check for huge phylogenies.