Cytosolic acetyl-coenzyme A is a signalling metabolite to control mitophagy
Summary
This Nature study identifies cytosolic acetyl-coenzyme A (AcCoA) as a direct signalling ligand for the mitochondrial mitophagy receptor NLRX1. The authors show that reductions in cytosolic (but not mitochondrial) AcCoA — caused by fasting, inhibition of ACLY or SLC25A1, mitochondrial protein import stress or treatment with KRAS inhibitors — trigger a selective, NLRX1-dependent mitophagy pathway. Mechanistically, AcCoA binds the LRR domain of NLRX1 and stabilises an autoinhibited NLRX1 conformation; when cytosolic AcCoA falls, NLRX1 oligomerises, engages LC3 via its LIR motif and drives autophagic removal of mitochondria. The work is validated across cell lines and in vivo mouse tissues and links mitophagy to therapeutic resistance to KRAS inhibitors in pancreatic cancer models.
Key Points
- Cytosolic AcCoA is a signalling metabolite that controls selective mitophagy rather than a mere metabolic substrate.
- NLRX1 is the principal mitophagy receptor sensing AcCoA via a conserved pocket in its LRR domain.
- AcCoA binding maintains NLRX1 autoinhibition (LRR–NACHT interaction); low AcCoA releases inhibition, promoting NLRX1 oligomerisation and LC3 recruitment.
- Fasting, ACLY or SLC25A1 inhibition, MPIS and KRAS inhibitors lower cytosolic AcCoA and induce NLRX1-dependent mitophagy in cells and mice.
- Supplementing acetate restores cytosolic AcCoA and blocks the mitophagy response, confirming causality.
- KRAS inhibitor treatment lowers ACLY/AcCoA and triggers mitophagy that helps tumours resist therapy; NLRX1 loss or mitophagy inhibition sensitises tumours to KRAS inhibitors.
Content summary
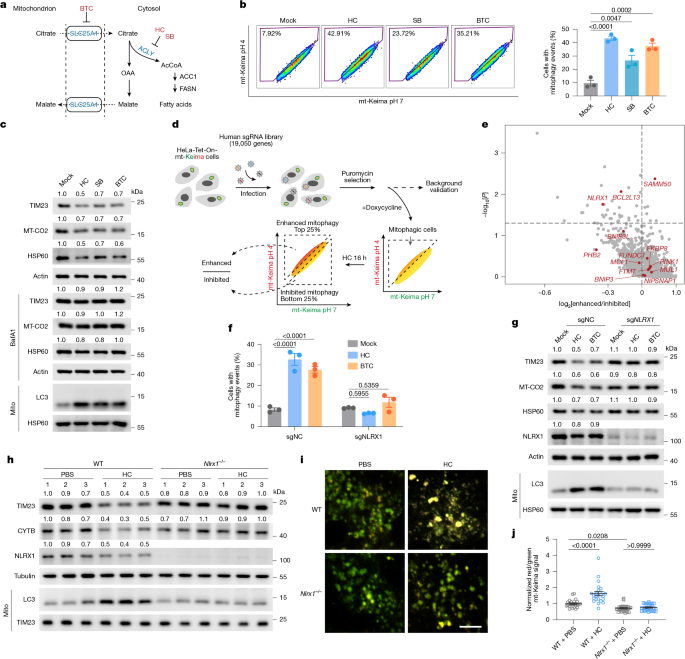
The authors begin by modelling mild nutrient restriction that mimics fasting (5 mM glucose, 2 mM glutamine), which triggers mitophagy in several cell lines. Metabolomics show a selective drop in cytosolic AcCoA in mitophagy-responsive cells. Pharmacological or genetic disruption of cytosolic AcCoA production (ACLY, SLC25A1, ACSS2) lowers cytosolic AcCoA and induces autophagic mitochondrial clearance; acetate rescues both AcCoA levels and blocks mitophagy.
A genome-wide CRISPR screen for factors required for AcCoA-reduction-induced mitophagy singled out NLRX1 as the top mitochondrial receptor. NLRX1 deficiency prevents mitophagy after AcCoA decline both in vitro and in vivo, while a mutant NLRX1 that cannot bind AcCoA (LRR 4A) is constitutively active and drives mitophagy. Biochemical assays, docking and purified-protein binding show direct, relatively high-affinity AcCoA binding (Kd ~6.6 μM) to a conserved pocket in the LRR domain; CoASH binds much more weakly. AcCoA stabilises the LRR–NACHT interaction and keeps NLRX1 monomeric; loss of AcCoA promotes oligomerisation and LC3 engagement via the LIR motif.
Importantly, the pathway operates independently of AMPK and mTOR signalling and does not require PINK1–Parkin. In mouse tissues, fasting decreases cytosolic AcCoA and increases mitophagy in brain and muscle but not liver; acetate or ACLY inhibition in vivo similarly modulates mitophagy. Finally, KRAS inhibitors reduce ACLY and cytosolic AcCoA, triggering NLRX1-dependent mitophagy that lowers oxidative stress and promotes tumour cell survival; blocking NLRX1 or mitophagy increases ROS and sensitivity to KRAS inhibition.
Context and relevance
This study provides a unifying metabolic signal — cytosolic AcCoA — that links nutrient status and mitochondrial protein-import stress to selective mitophagy through direct sensing by an innate-immune family receptor. It expands the functional repertoire of NLR proteins from immune sensing to metabolite sensing and selective organelle quality control. For cancer biology, the finding that KRAS inhibitors downregulate ACLY/AcCoA and thereby activate protective mitophagy reveals a metabolic resistance mechanism that is targetable: combining KRAS inhibitors with mitophagy blockers or strategies that restore cytosolic AcCoA may improve therapeutic efficacy.
Author style
Punchy: this paper nails a neat molecular mechanism — AcCoA as a ligand — and links it to physiologically relevant processes (fasting) and a clinically urgent problem (KRAS inhibitor resistance). Read the data if you care about metabolism ↔ autophagy cross-talk or developing combination cancer therapies.
Why should I read this?
Want the short version: AcCoA isn’t just fuel — it tells a mitophagy receptor what to do. If you’re into metabolism, autophagy, mitochondrial quality control or resistance mechanisms in cancer, this saves you time: the authors pin down the sensor (NLRX1), the ligand (cytosolic AcCoA), the mechanism (binding → oligomerisation → LC3 recruitment) and show it matters in animals and in KRAS-driven tumours. Handy if you plan experiments or treatments that tinker with ACLY, acetate metabolism or mitophagy.