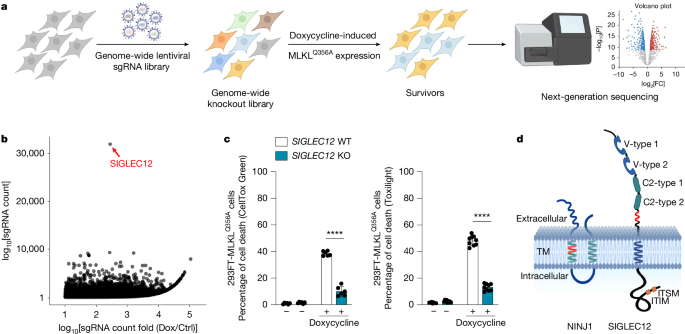
SIGLEC12 mediates plasma membrane rupture during necroptotic cell death
Article Date: 12 November 2025
Article URL: https://www.nature.com/articles/s41586-025-09741-1
Article Image: Figure 1
{kind=link}
Summary
This Nature study used a genome-wide CRISPR–Cas9 screen and follow-up experiments to identify SIGLEC12 as a critical mediator of plasma membrane rupture (PMR) during necroptosis downstream of MLKL activation. Knockout or knockdown of SIGLEC12 prevents PMR (measured by CellToxGreen staining and LDH release) without preventing the upstream activation of RIPK1/RIPK3/MLKL or loss of cellular ATP, resulting in a characteristic swollen ‘bubble’ morphology and reduced release of cytosolic inflammatory mediators. SIGLEC12 shares a short homology motif with NINJ1 (the NISI motif), forms puncta and fibrils during necroptosis, and is cleaved extracellularly to produce a ~20 kDa fragment that contains the NISI motif and is sufficient to drive PMR. The transmembrane serine protease TMPRSS4 mediates cleavage at Arg410 to activate SIGLEC12. Several cancer-associated and population variants alter SIGLEC12 cleavage or function. Importantly, the SIGLEC12 requirement for PMR appears to be human-specific: mouse cells do not require Siglec12 for necroptotic PMR, reflecting sequence divergence and species differences in SIGLEC family members. The work positions SIGLEC12 and TMPRSS4 as potential druggable targets to modulate proinflammatory necroptotic death in humans.
Author note (punchy)
Short version: they found the molecular switch that rips open human cells during necroptosis. If you’re interested in cell-death mechanisms, inflammation or therapeutic blocking of damaging lytic death, this is a major find — especially because it looks to be human-specific.
Key Points
- A genome-wide CRISPR screen in MLKL-activated human cells flagged SIGLEC12 as the top hit protecting cells from MLKL-driven death when knocked out.
- SIGLEC12 loss prevents plasma membrane rupture (PMR) during necroptosis but does not stop MLKL oligomerisation or upstream kinase activation — cells still lose ATP.
- SIGLEC12 forms puncta and fibrils and contains a short NINJ1/SIGLEC12 homology motif (NISI) required for PMR activity.
- SIGLEC12 is cleaved during necroptosis to produce a ~20 kDa extracellular fragment (containing the NISI motif) that is sufficient to induce PMR when expressed as a cleavage-mimetic truncation.
- The transmembrane serine protease TMPRSS4 cleaves SIGLEC12 at Arg410; TMPRSS4 knockdown or protease-dead mutants block cleavage and PMR.
- Cancer-associated and population SIGLEC12 variants (for example S458F, R528W) can impair TMPRSS4-mediated cleavage and thus PMR, with potential implications for cancer biology and infection susceptibility.
- SIGLEC12-dependent PMR appears to be species-specific (likely human-specific): mouse Siglec12 does not recapitulate the requirement, reflecting evolutionary divergence in SIGLEC sequences.
- Because SIGLEC12 regulates release of inflammatory mediators (HMGB1, IL-1β, IL-8, CXCL1, TNF), modulating its activation could tune necroptosis-driven inflammation in disease.
Context and relevance
Necroptosis is an inflammatory, lytic form of programmed cell death driven by MLKL. Previously, NINJ1 was shown to mediate PMR in several lytic death pathways, but not downstream of MLKL in necroptosis. This paper fills that gap by describing a distinct mechanism in human cells: SIGLEC12 acts downstream of MLKL to execute PMR after proteolytic activation by TMPRSS4. The species-specific element is significant — it warns that mouse models may miss human-relevant steps in necroptosis and highlights SIGLEC12/TMPRSS4 as more translationally relevant targets for therapies aimed at controlling damaging necroptosis in human diseases (for example inflammatory conditions, viral infections or cancer microenvironments where necroptosis is engaged).
Why should I read this?
Because it actually changes how we think necroptosis rips cells apart — and it might explain why some therapies that work in mice stumble in humans. The paper gives you a clear molecular chain: MLKL activation → SIGLEC12 cleavage by TMPRSS4 → fibril formation → plasma membrane rupture. If you work on inflammation, programmed cell death, cancer immunology or drug targets for lytic cell death, this saves you hours of digging and points straight at two druggable proteins.
Source
Original article: SIGLEC12 mediates plasma membrane rupture during necroptotic cell death — Nature (12 Nov 2025)