Decay of driver mutations shapes the landscape of intestinal transformation
Article meta
Article Date: 03 December 2025
Article URL: https://www.nature.com/articles/s41586-025-09762-w
Article Image: Figure 1 (Nature)
{kind=link}
Summary
This Nature study uses engineered mouse intestinal fields and ENU mutagenesis to show that the survival and selection of classic colorectal-driver mutations (principally Apc and Ctnnb1/β-catenin) depend strongly on the pre-existing mutational or functional ‘priming’ state of the epithelium. Different priming events (for example, KrasG12D, Trp53 loss, Fbxw7 loss, Pten loss or Apc heterozygosity) change both the rate of tumour formation and which specific mutations are favoured. Critically, many conditional driver mutations are lost over time in unprimed tissue — a phenomenon the authors term “decay” — and the probability that a given mutation survives to drive a tumour varies with its protein position and with the order of events. Human cohort analyses (AACR GENIE and INCISE polyp data) show parallel shifts in APC truncation patterns associated with KRAS status, suggesting the mouse findings are relevant to human colorectal cancer evolution.
Key Points
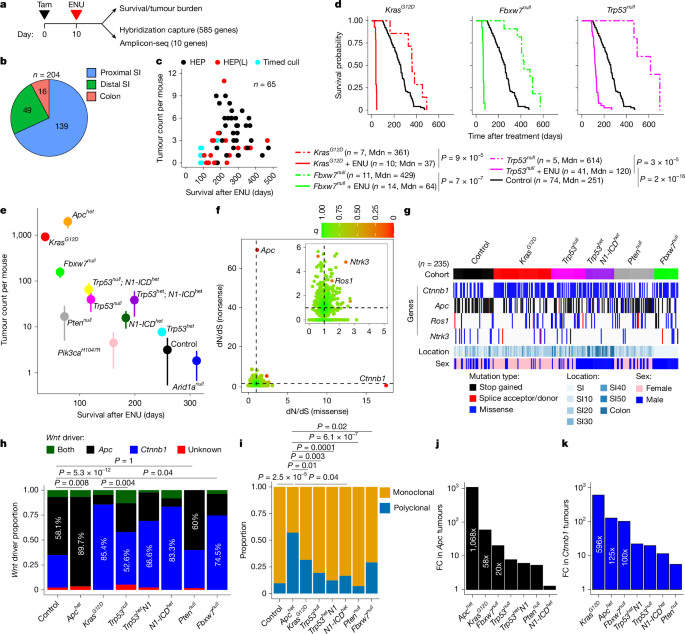
- Priming the intestinal epithelium with specific driver alterations (eg. KrasG12D, Trp53null, Fbxw7null, Ptennull, Apchet) dramatically alters susceptibility to ENU-driven transformation and tumour multiplicity.
- Apc truncations and Ctnnb1 exon-3 missense changes are the dominant drivers in this ENU model; however, which exact codons/truncation sites are selected depends on the priming context.
- Rescue (ENU then tamoxifen) experiments demonstrate pronounced negative selection (decay) of many conditional driver mutations when the permissive field is absent at the time mutations arise — i.e. many potential driver clones fail to survive long enough to form tumours unless the tissue is primed.
- Different Ctnnb1 exon-3 mutations and APC truncation bins show distinct observed/expected ratios: some are over-represented because of mutation bias and positive selection, others are depleted by negative selection.
- Order and epistasis matter: prior or subsequent loss of genes such as FBXW7 or activation of KRAS can either protect against or promote transformation from specific APC/CTNNB1 mutations.
- Human CRC data support the concept of priming: KRAS-mutant tumours more often retain additional APC 20–amino-acid repeats, consistent with a different selection landscape for APC truncations in KRAS-primed tissue.
Content summary
The authors created multiple Villin-CreERT2 mouse lines carrying conditional oncogenic or loss-of-function alleles (KrasG12D, Trp53, Fbxw7, Pten, Arid1a, Pik3ca, Notch1 and Apc heterozygosity). They exposed these primed fields (tamoxifen first) to the alkylating mutagen ENU, then sequenced hundreds of tumours across cohorts using a targeted hybrid capture panel and deep amplicon sequencing.
Key experimental observations:
– KrasG12D priming produced an extreme increase in tumour multiplicity and skewed drivers towards Ctnnb1 changes; other priming genotypes had intermediate effects.
– Ctnnb1 exon-3 driver mutations clustered in a handful of codons; their distribution reflected both the underlying ENU trinucleotide mutation spectrum and selection by the priming context.
– APC truncations were analysed by dividing the protein into spatial bins (A–E). Selection biases varied by priming: some contexts disfavoured N‑terminal truncations, others selected for truncations that retain specific domains (eg. Armadillo repeats or 20‑aa repeats).
– Rescue (ENU→tamoxifen) experiments showed that many conditional driver mutations decay (are lost) when priming is applied after mutation induction; the decay rate depended on mutation identity and priming genotype, revealing negative selection against many single-allele driver events in otherwise unprimed tissue.
Finally, analysis of ~9,000 GENIE CRC samples and ~810 polyps from the INCISE cohort revealed shifts in APC truncation patterns associated with KRAS status and with relative variant allele frequencies, consistent with priming and mutational order influencing human colorectal tumour evolution.
Context and relevance
Why this matters: the paper reframes why APC loss is ubiquitous in colorectal cancer. It shows that the capability of WNT-pathway mutations to produce a spectrum of fitness outcomes — from lethal to highly transforming — lets them act as flexible drivers across diverse predisposing fields. That explains both why APC is so often involved and why the precise APC or CTNNB1 lesion differs between tumours. The experiments also highlight that many potential driver clones never persist unless the microenvironment/epithelial field is permissive; therefore, detection of early driver mutations in normal tissue or polyps doesn’t automatically mean progression will occur — order and context are decisive.
Implications:
– For researchers: models of tumour initiation must account for priming, mutation order and negative selection (decay) to predict which clones will progress.
– For clinicians and biomarker development: mutation identity and allele frequency together with context (eg. KRAS status) may help stratify polyp risk.
– For therapeutics: interventions that alter the tissue field or block permissive epistatic interactions could reduce the chance that nascent driver clones survive.
Why should I read this?
Short and to the point: if you care about how colorectal cancers start and which early mutations actually matter, this paper is gold. The team read through a massive set of mouse experiments and human datasets to show that it isn’t just which mutation appears, but when and where it appears — and whether the tissue is already ‘primed’ — that determines if a clone survives to form a tumour. Saves you time: they unpack the messy reality behind APC/CTNNB1 selection, mutation bias and clonal survival so you don’t have to wade through dozens of smaller contradictory studies yourself.
Author style
Punchy: This is a high-impact, mechanistic tour-de-force. The findings force us to rethink simple linear models of colorectal tumour initiation and highlight selection dynamics (including powerful negative selection) that have been underappreciated. If you work on tumour evolution, early detection, or CRC risk stratification, the detailed data and methods here are worth studying closely.