Somatic evolution following cancer treatment in normal tissue
Summary
This Nature study used ultra-sensitive duplex sequencing on 168 cancer-free samples (16 tissue types) and blood from 22 heavily pre-treated patients to map how cancer therapies and lifestyle exposures reshape somatic mutations and selection in normal tissues. The team identified therapy-specific mutational signatures (notably platinum, temozolomide and radiation footprints), tissue-dependent mutation burdens (liver high; brain/pituitary low), and evidence that treatments both induce mutations and act as selection pressures — for example enriching for drivers such as TP53, PPM1D and CHEK2. The work quantifies how a course of therapy can be equivalent to many years of ageing in mutational load, and shows immunotherapy can select for particular clones even if it is not mutagenic itself.
Key Points
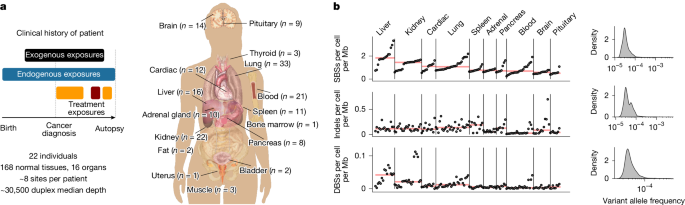
- Duplex sequencing (82.5 kb panel) on 156 normal tissue biopsies + blood revealed >166k single-base substitutions and therapy-linked signatures in normal tissues.
- 16 distinct single-base substitution (SBS) signatures were found; some matched COSMIC (e.g. SBS31/35 for platinum), others were novel (SBS-A–J).
- Platinum chemotherapy leaves a measurable mutational footprint across tissues; blood showed the highest mutations per platinum cycle.
- Temozolomide, alkylators and radiotherapy also produce distinct signatures in normal tissue; temozolomide footprint (SBS-F) was common in treated patients.
- Tissue and exposure shape mutation rates: liver and lung accrue many exogenous mutations (alcohol, smoking), brain shows low exogenous contribution.
- Positive selection is evident in normal tissue for classic cancer genes, particularly TP53; selection patterns vary by tissue and treatment.
- Immunotherapy did not generally increase mutation counts but was associated with selection for clones carrying drivers (e.g. TP53, PPM1D, B2M).
- Treatment can create a punctuated burst of mutagenesis equivalent to years of ageing (e.g. 6 platinum cycles ~ many ageing years depending on tissue).
Content summary
The authors accessed the PEACE autopsy resource, sequencing small frozen biopsies from 16 normal tissues and blood from 22 patients with detailed treatment histories. Using duplex sequencing they minimised artefacts and measured ultra-rare variants, then performed de novo mutational-signature extraction (hierarchical Dirichlet processes) and dN/dS-based selection analyses adjusted for coverage and context.
They linked specific mutational footprints to platinum agents (SBS31/35), temozolomide (SBS-F), radiation-associated indels (ID-E) and other agents, quantifying per-tissue mutation rates per treatment cycle. Smoking and alcohol exposures produced tissue-specific signatures (SBS4 and SBS-B respectively). Importantly, many driver mutations in normal tissue could be probabilistically attributed to treatment-related signatures, although not all treatment-induced mutations showed evidence of positive selection.
Context and relevance
This study provides concrete, quantitative evidence that cancer therapies — widely used and life-saving — also leave measurable genomic scars in histologically normal tissues. It brings together mutagenesis and selection: therapies both generate mutations and change the competitive landscape so particular clones expand. The findings are relevant to clinicians, researchers and survivorship planning because they help explain long-term risks (second malignancies, clonal haematopoiesis) and show that risk varies by tissue, treatment type and dose.
Why should I read this?
Because it answers the obvious but underexplored question: what does cancer treatment do to the rest of your body’s cells? If you want the fast take — treatments can hit normal tissue hard, leave distinct mutational fingerprints and push some cell clones to expand. The paper gives hard numbers (mutations per cycle, tissue differences) that matter for research, follow-up strategies and weighing long-term risks versus benefit.
Source
Article Date: 10 December 2025