An integrated view of the structure and function of the human 4D nucleome
Summary
Article Date: 17 December 2025
Article URL: https://www.nature.com/articles/s41586-025-09890-3
Article Title: An integrated view of the structure and function of the human 4D nucleome
Article Image: Figure 1
{kind=link}
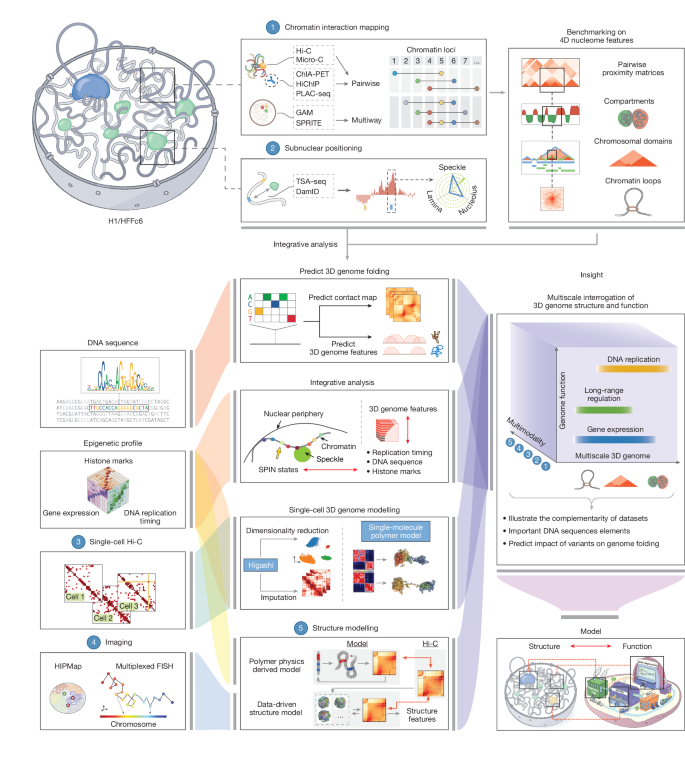
This Nature consortium paper from the 4D Nucleome Project presents an extensive, integrated resource describing how the human genome is folded and how that folding links to function. The authors benchmark and combine many genomic assays (Hi-C, Micro-C, SPRITE, GAM, ChIA-PET, PLAC-seq, TSA–seq, DamID, iMARGI and single-cell methods) in two reference cell types (H1 embryonic stem cells and HFFc6 fibroblasts), produce a union catalogue of >140,000 loops per cell type, define nine SPIN spatial states that map loci relative to nuclear bodies, and generate populations of 1,000 single-cell 3D genome models. The study links structure to replication timing, transcription, enhancer connectivity and cell-to-cell variability, and demonstrates predictive sequence-based models for folding (Akita-style). All datasets and code are publicly available via the 4D Nucleome data portal.
Key Points
- Comprehensive benchmarking: multiple 3D genome assays were systematically compared; each captures distinct and complementary features (compartments, TAD boundaries, loops or large-scale co-associations).
- SPIN annotation: integration of TSA–seq, DamID and Hi-C yields nine reproducible SPIN states (speckle, interior active/repressive states, near-lamina and lamina) that stratify replication timing, histone marks and RNA association.
- Large loop catalogue: by combining platforms the consortium assembled a union set of ~140–146k loops per cell type; no single assay finds them all and different methods preferentially detect different loop classes.
- 3D single-cell models: integrative genome modelling produced populations of 1,000 single-cell structures revealing cell-to-cell variability and predicting distances to nuclear bodies (speckles, nucleoli, lamina) that correlate with expression changes.
- Structure–function links: genes with more distal enhancer contacts tend to be more highly and broadly expressed; housekeeping genes frequently engage many distal enhancers but in cell-type-specific ways.
- Method guidance: SPRITE and Hi-C best detect compartmentalisation (small SPRITE clusters emphasise compartments); Micro-C excels at structural loops; ChIA-PET/PLAC-seq enrich for transcription-related loops; GAM and SPRITE better capture loci co-associating around large nuclear bodies.
- Single-cell variability: loops, TAD-like domains and compartments exist at the single-cell level but are variable; loop formation is stronger when anchors lie in the same TAD or compartment in that cell.
- Predictive models: sequence-based deep-learning models trained on Micro-C can forecast folding changes from mutations and identify candidate sequence determinants and TF motifs linked to cell-type-specific boundaries.
- Resource and reproducibility: all raw data, annotations, models and analysis code are publicly available at the 4D Nucleome Data Coordination and Integration Center (https://data.4dnucleome.org/) and linked Zenodo repositories.
Why should I read this?
Short version: if you care about how genome folding affects gene control, this is the atlas. It saves you the massive effort of reading dozens of separate method papers — the consortium benchmarked them, produced a huge loop catalogue, created single-cell 3D model populations, and tied structure to replication and transcription. Read it if you want a practical guide to which assay to use, a big set of validated structural annotations (SPIN, loops, TADs) or sequence-based predictions of folding effects. In other words — excellent value for time spent.