GlycoRNA complexed with heparan sulfate regulates VEGF-A signalling
Summary
This paper shows that glycoRNAs (sialylated N-glycan–modified small RNAs) and specific cell-surface RNA-binding proteins (csRBPs) assemble into clustered RNP domains on the cell surface that are nucleated by heparan sulfate proteoglycans (HSPGs). Formation of these csRNP clusters requires intact heparan sulfate (HS) chains, specifically N-sulfation and 6-O-sulfation. The canonical HS-binding C‑terminal domain of VEGF‑A165 binds glycoRNAs at these csRNP sites and, unexpectedly, this interaction limits VEGF‑A165 signalling through VEGFR2 (reduced ERK activation). Removing cell-surface RNA with RNases or creating an arginine-to-lysine (R/K) mutant in the VEGF‑A165 HS domain (which preserves HS binding but reduces RNA binding) increases VEGF‑A165 cell binding, VEGFR2/ERK activation and angiogenic behaviour in 2D, 3D and in vivo models (mouse retina and zebrafish). The authors therefore nominate csRNPs as a novel, HS-dependent extracellular brake on HS-binding growth factor activity.
Key Points
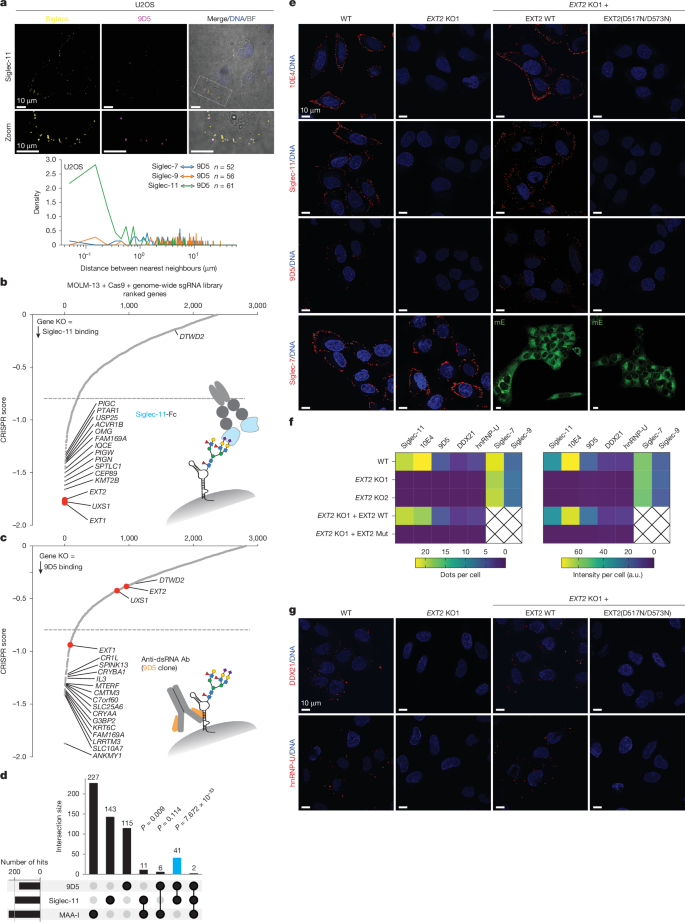
- Cell-surface glycoRNAs and csRBPs form nanoscale, HS-dependent clusters (csRNPs) tethered by HSPGs; intact HS polymers are required.
- N‑sulfation and 6‑O‑sulfation of HS specifically promote csRNP assembly; genetic knockouts or enzymatic removal of these modifications disrupt clusters.
- Siglec‑11 is an RNA-sensitive cell-surface lectin that colocalises with glycoRNAs and captures sialoglycoRNAs in vitro.
- VEGF‑A165 (but not VEGF‑A121) binds directly to small RNAs at the cell surface via its arginine-rich HS-binding C‑terminus; this binding antagonises VEGF‑A165 signalling.
- RNase treatment or mutation of arginines to lysines in the HS domain (VEGF‑A165 HS(R/K)) increases VEGF‑A165 binding to cells, VEGFR2 phosphorylation and downstream ERK activation.
- Enhanced VEGF activity from RNA removal or the R/K mutant drives greater endothelial migration and vessel‑like structure formation in microfluidic 3D assays and increases angiogenesis in mouse retina and zebrafish embryos.
- The R/K mutant retains HS binding but loses glycoRNA binding, providing a tool to separate HS-dependent tethering from RNA-dependent antagonism.
- Findings reveal a new extracellular regulatory layer: csRNPs modulate growth-factor bioavailability and signalling, with implications for development and disease.
Context and relevance
This is an advance in extracellular matrix and RNA biology that links glycobiology, cell-surface RNA, and canonical growth-factor signalling. VEGF‑A165 is a fundamental pro‑angiogenic factor; showing that cell-surface glycoRNAs act as negative modulators of VEGF‑A165 reframes how extracellular cues tune angiogenesis. The mechanism (HS-dependent csRNP clustering + selective RNA binding by arginine residues) is likely relevant to other HS-binding growth factors and to disease contexts where HS structure or extracellular RNA abundance changes (tumour microenvironments, wound healing, vascular disorders).
Why should I read this?
Short version: it’s clever and matters. If you work on angiogenesis, extracellular matrix signalling, RNA biology or therapeutic targeting of VEGF, this paper saves you the time of digging through dozens of disparate studies — it brings glycoRNAs, HS chemistry and VEGF biology together and shows a clear functional effect in cells and animals. Think of csRNPs as a tunable brake on HS-binding growth factors — a neat conceptual and experimental hook with real in vivo evidence.
Author style
Punchy — the authors combine genetics, biochemistry, imaging and in vivo models to make a crisp mechanistic claim. Experiments are directed to show cause and effect (enzymes, KO/rescue, RNase, mutant VEGF, microfluidics, mouse and zebrafish), so the conclusion feels well supported and immediately relevant.