Population-scale sequencing resolves determinants of persistent EBV DNA
Summary
Article Date = 28 January 2026
Article URL = https://www.nature.com/articles/s41586-025-10020-2
Article Image = https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41586-025-10020-2/MediaObjects/41586_2025_10020_Fig1_HTML.png
Key Points
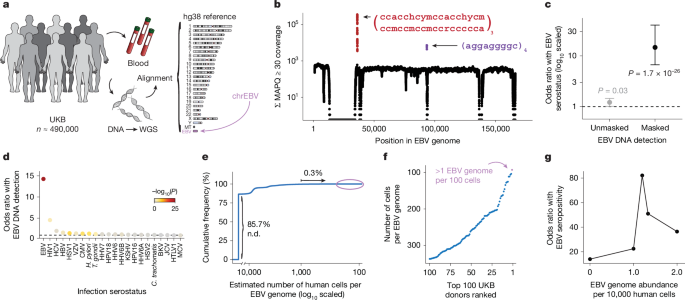
- The authors developed a scalable pipeline to recover and quantify Epstein–Barr virus (EBV) DNA from blood whole-genome sequencing (WGS) data by extracting reads mapped to the EBV contig in hg38 and masking low-complexity, biased regions.
- In UK Biobank (n≈490k) and All of Us (n≈245k) cohorts, 9.7–11.9% of donors had detectable EBV DNAemia (≥1.2 genomes per 10,000 cells), reflecting a tail of individuals with higher circulating EBV DNA.
- EBV DNAemia associates with many clinical phenotypes (PheWAS), including known links (Hodgkin lymphoma, splenic disease, rheumatoid arthritis, COPD) and suggested links (fatigue/ME/CFS, cardiovascular and renal conditions).
- GWAS and ExWAS identified 22 genome-wide significant loci for EBV DNAemia; strongest signals map to the MHC/HLA region and immune-regulatory genes (CTLA4, EOMES, PTPN22, SLAMF7).
- Single-cell and ATAC analyses show enrichment of associated genes in B cells and antigen-presenting cells; pathway enrichment implicates antigen processing and presentation (MHC), and T-cell regulation.
- HLA allele-level analyses and NetMHC predictions indicate that stronger predicted presentation of EBV peptides—particularly via MHC class II—correlates with lower likelihood of persistent EBV DNA in blood.
- Aggregated viral reads permit assessment of EBV genomic diversity across populations; many EBV tumour-associated variants appear common in healthy cohorts, suggesting geographic drift rather than sole pathogenicity.
- The approach repurposes existing population-scale WGS to create a new molecular biomarker (EBV DNAemia) and demonstrates host genetics shape lifelong viral persistence.
Content summary
This study shows that routinely generated blood WGS contains usable EBV reads when aligned to an EBV contig in the human reference. By masking problematic repetitive regions and normalising coverage, the team computed per-person EBV DNA copy estimates across nearly half a million UK Biobank participants and independently in All of Us.
They defined a threshold (≈1.2 EBV genomes per 10,000 cells) to call EBV DNAemia and used that binary trait for phenome-wide scans and genetic analyses. The PheWAS recovered expected associations (lymphoproliferative and autoimmune phenotypes) and nominated additional clinical links. GWAS/ExWAS uncovered multiple loci—most prominently at the HLA/MHC—implicating antigen presentation pathways and B-cell biology in controlling persistent EBV DNA.
Integrating four-digit HLA calls with peptide-binding predictions (NetMHCpan/NetMHCIIpan) showed that individuals whose HLA alleles are predicted to present EBV peptides more effectively—especially via class II—are less likely to have persistent EBV DNA in blood. Aggregating viral sequences across cohorts also revealed that many EBV variants previously reported from tumours are common in healthy people, arguing for geographic structure or additional factors for pathogenicity.
Context and relevance
Why this matters: EBV infects >90% of adults and underlies cancers and autoimmune associations, yet why some people carry more circulating viral DNA has been unclear. This paper demonstrates that inter-individual differences in viral persistence are partly genetic, driven by antigen-presentation and immune-regulatory loci. The methods unlock retrospective virome measurement from existing WGS datasets, enabling powerful, population-scale host–virus studies without fresh assays.
Relevance to ongoing trends: the work sits at the intersection of biobanking, population genomics and infectious disease — repurposing WGS for virome surveillance is timely as biobanks expand globally. It also connects genetic risk for autoimmune disease with differential viral persistence, informing hypotheses about viral triggers of autoimmunity and opportunities for targeted immunological follow-up.
Author style
Punchy: big cohorts, clever re-use of “discarded” reads, and rigorous genetic follow-up. This paper is a neat demonstration that population WGS can do double duty — genome and virome — and that MHC-driven antigen presentation strength is a key determinant of who carries detectable EBV DNA. If you care about infectious triggers of immune disease, this one sharpens the tools and the questions.
Why should I read this?
Short answer: because it’s clever and useful. The team squeezed new biology out of existing WGS by pulling EBV reads that are normally ignored, then linked persistence to human genes (especially HLA) and clinical outcomes. If you work in genomics, immunology, virology, or biobank science, this saves you time — it tells you how to measure EBV at population scale and why those measurements map back to immune genes and disease. Plus, it opens ways to study other persistent viruses without new wet-lab assays.