
Activated ATF6α is a hepatic tumour driver restricting immunosurveillance
Article Date: 04 February 2026
Article URL: https://www.nature.com/articles/s41586-025-10036-8
Summary
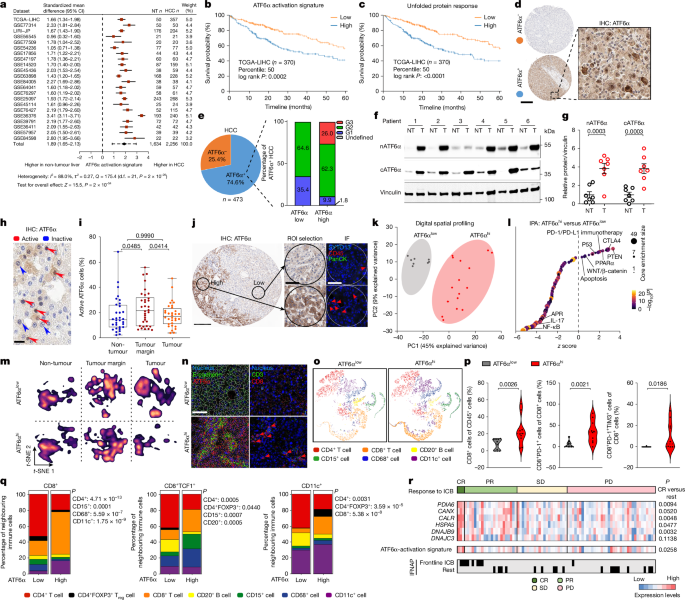
This Nature paper identifies chronically activated ATF6α (the cleaved, nuclear form nATF6α) as a key driver of hepatocellular carcinoma (HCC). The authors combine human tumour analyses with multiple mouse genetic and intervention models to show that sustained hepatocyte ATF6α activation represses the gluconeogenic enzyme FBP1, rewires glucose metabolism towards glycolysis, depletes local glycogen/glucose, increases lactate, and creates a metabolically hostile microenvironment that impairs CD8+ T cell function and promotes T cell exhaustion. Hepatocyte ATF6α activation is sufficient to induce spontaneous HCC in mice; conversely, Atf6 deletion or hepatocyte-targeted antisense oligonucleotides (GalNAc‑ASO) reduce ER stress, restore FBP1, mitigate tumour development and lower immunosuppression. Notably, ATF6α-high tumours display features that make them more responsive to PD-1 blockade in preclinical models and in a human cohort of anti-PD-1-treated patients.
Key Points
- Activated (nuclear) ATF6α is enriched in human HCC, correlates with aggressive grade and reduced survival, and defines a distinct UPR-linked signature.
- nATF6α directly represses the Fbp1 promoter, shifting hepatocyte metabolism from gluconeogenesis to glycolysis, depleting glycogen/glucose and increasing lactate output.
- Hepatocyte-specific chronic ATF6α activation causes ER stress, DNA damage, compensatory proliferation and, ultimately, spontaneous HCC in mice.
- Restoring catalytically active FBP1 reverses metabolic reprogramming, reduces ER stress and lowers tumour burden; catalytically dead FBP1 does not rescue the phenotype.
- Genetic deletion of Atf6 or hepatocyte-targeted GalNAc‑ASO knockdown reduces ER stress, preserves FBP1, and markedly limits HCC incidence in several preclinical models.
- ATF6α-driven tumours are immunosuppressive (more exhausted CD8+PD‑1+ T cells, M-MDSCs, PD‑L1+ macrophages) but form immune niches that render them paradoxically more responsive to PD‑1 blockade; anti‑PD‑1 therapy reduced tumour burden and improved T cell glycolytic activity in mouse models with ATF6α activation.
Content summary
The authors first show increased ATF6/ATF6α-activation signatures in 22 human HCC datasets and link high ATF6α activity to worse survival and aggressive molecular subtypes. Spatial transcriptomics, proteomics and imaging mass cytometry reveal ATF6α-high regions are hypoxic, glycolytic and immune-exhausted with reduced FBP1.
They generated hepatocyte-specific nATF6α transgenic mice and AAV-mediated models. Persistent ATF6α activation produced ER stress, suppressed FBP1 transcription (CUT&RUN/ATAC data show direct promoter binding and chromatin closure), increased glycolytic enzyme expression, depleted glycogen/glucose and drove liver injury, DNA damage and proliferation—progressing to spontaneous HCC with chromosomal aberrations similar to human tumours.
Mechanistically, restoring FBP1 in hepatocytes reversed many metabolic and injury phenotypes and reduced tumour formation. Conversely, deleting Atf6 (global or hepatocyte-specific) or treating mice with GalNAc‑ASO against Atf6 lowered ER stress, preserved FBP1, reduced glycolysis and decreased tumour incidence across multiple dietary and oncogene-driven models.
Immune profiling (scRNA-seq, IMC, MIBI) shows ATF6α activation creates a glucose‑poor, lactate‑rich microenvironment that impairs CD8+ T cell glycolysis and function, promoting exhaustion. Yet tumours with this phenotype formed immune niches (CD8+ with CD11c+ DCs and CD4+ help) and were more sensitive to PD‑1 blockade; anti‑PD‑1 therapy increased LDH and glycolytic activity in tumour CD8+ T cells and reduced tumour burden. Genetic PD‑1 deletion similarly improved tumour control in ATF6α-driven mice.
Context and relevance
This study links chronic unfolded protein response signalling (via ATF6α) to metabolic reprogramming that both drives hepatocyte transformation and actively suppresses anti‑tumour immunity. It connects UPR biology, tumour metabolism (FBP1 loss → glycolysis), immune exhaustion and immunotherapy response in HCC. Clinically, ATF6α activity could be a dual biomarker: prognostic for aggressive disease and predictive of better response to PD‑1 blockade in specific contexts. Therapeutically, hepatocyte‑targeted strategies that prevent ATF6α activation or restore FBP1 may reduce tumour initiation and improve immune surveillance.
Author style
Punchy: this is a mechanistic tour de force — ATF6α isn’t just a stress marker, it drives liver cancer by starving T cells of fuel. The data are consistent across human cohorts and robust mouse models, and they point to actionable interventions (FBP1 restoration, ATF6 targeting with GalNAc‑ASO, or refining patient selection for ICB).
Why should I read this
Quick answer — if you work in liver cancer, tumour immunology or tumour metabolism, this paper explains a neat causal chain: chronic ATF6α → FBP1 down → glycolysis + lactate → T cell dysfunction → HCC. It tells you why some HCCs look exhausted but still respond to PD‑1, and suggests real therapeutic angles. We’ve done the heavy reading for you; skim the figures for CUT&RUN, the mouse survival curves and the PD‑1 treatment data.