
CLCC1 governs ER bilayer equilibration to maintain lipid homeostasis
Article metadata
Article Date: 25 February 2026
Article URL: https://www.nature.com/articles/s41586-026-10161-y
Article Image: Figure 1 (PNG)
{kind=link}
Summary
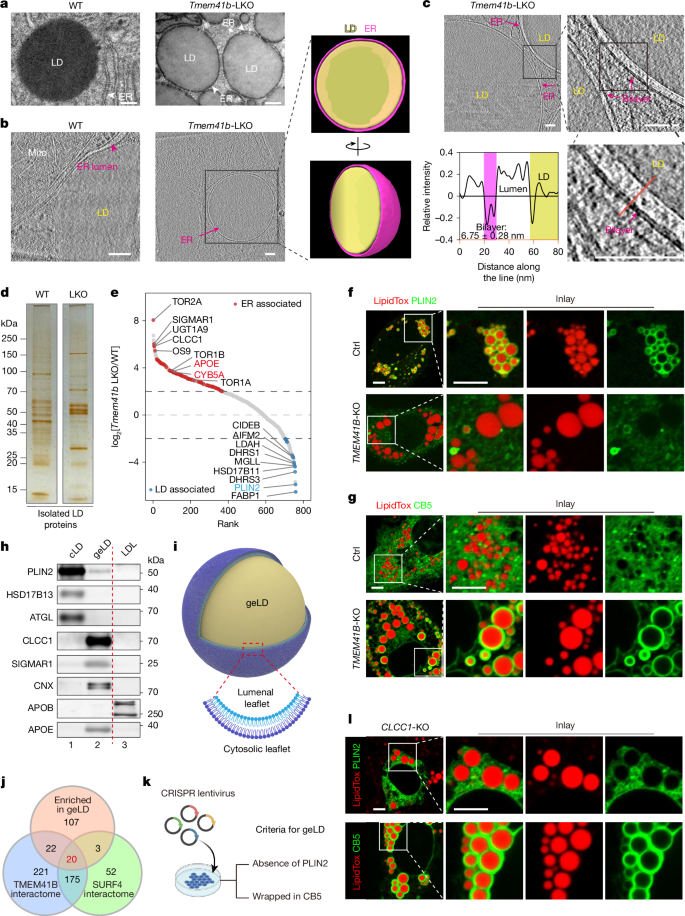
This Nature study identifies CLCC1 as a critical regulator of endoplasmic reticulum (ER) bilayer equilibration and lipid homeostasis. The authors show that defective phospholipid scrambling (previously linked to TMEM41B) produces giant ER-enclosed lipid droplets (geLDs) with imbalanced leaflets. CLCC1 is enriched on these curved, lipid-enclosing ER membranes, forms oligomers, biochemically associates with TMEM41B and promotes TMEM41B-dependent phospholipid scrambling. Loss of CLCC1 in liver reproduces key TMEM41B-deficiency phenotypes: failure in APOB lipidation, near-abolition of circulating triglyceride-rich lipoproteins, accumulation of geLDs and rapid progression to metabolic-dysfunction-associated steatohepatitis (MASH). In vitro and cell assays indicate CLCC1 amplifies TMEM41B scramblase activity despite having no intrinsic scramblase function. The work connects membrane leaflet imbalance to lipoprotein biogenesis and metabolic disease susceptibility.
Key Points
- TMEM41B scramblase deficiency produces geLDs: large lipid droplets fully encased by a single ER bilayer with asymmetric leaflets.
- geLDs are compositionally distinct from conventional cytosolic LDs (cLDs): depleted of PLIN2 and APOB, enriched for specific ER proteins (e.g. torsins, sigma-1 receptor, cytochrome b5, APOE).
- CLCC1 was identified from geLD proteomes and interactomes; CRISPR loss of CLCC1 phenocopies TMEM41B deficiency and induces geLDs.
- CLCC1 physically associates with TMEM41B, oligomerises on curved ER membranes and recruits/stabilises TMEM41B at sites of leaflet imbalance.
- Cellular and in vitro assays show CLCC1 enhances TMEM41B-mediated phospholipid scrambling, although CLCC1 alone lacks scramblase activity.
- Acute hepatic CLCC1 inactivation depletes plasma triglycerides and VLDL/LDL, reduces APOB100, impairs VLDL secretion and quickly drives MASH-like pathology.
- CLCC1 levels fall in obese/high-fat models; re-expression of CLCC1 improves hepatic lipid accumulation and liver damage markers.
- Mathematical modelling and lipid quantification support a lumenal phospholipid shortfall on geLD surfaces that is stabilised by recruited lumenal proteins.
- MTP inhibition can redirect neutral lipids from geLDs to cLDs, linking leaflet imbalance to directional lipid transfer mechanisms.
Content summary
The paper uses cryo-electron tomography, TEM, proteomics, targeted CRISPR screening and biochemistry to map how ER leaflet imbalance arises and is handled. Loss of TMEM41B scramblase function causes phospholipids to accumulate on the cytosolic leaflet, producing highly curved ER bilayers that envelop large lipid droplets (geLDs). Proteomic analysis of geLDs flagged CLCC1, an understudied ER membrane protein, which the authors validated genetically and biochemically as a regulator of lipid scrambling.
CLCC1 forms oligomers and concentrates on curved ER membranes around geLDs. It co-purifies with TMEM41B and promotes recruitment/stabilisation of the scramblase to imbalanced leaflets. In cells, CLCC1 loss leads to cytosolic accumulation of newly synthesised phosphatidylcholine (a readout of impaired scrambling); in vitro reconstitution shows CLCC1–TMEM41B complexes increase TMEM41B scramblase efficiency compared with TMEM41B alone. Hepatic CRISPR inactivation of CLCC1 causes loss of circulating triglyceride-rich lipoproteins, failure of APOB lipidation, and rapid liver pathology resembling MASH. Conversely, CLCC1 expression mitigates steatosis in obese mice. The study proposes a homeostatic circuit in which CLCC1 senses or binds imbalanced, curved ER bilayers and amplifies scramblase activity to restore leaflet equilibrium and permit VLDL assembly.
Context and relevance
This work addresses a fundamental cell-biology gap: how asymmetrically synthesised phospholipids cross the ER bilayer to support organelle biogenesis and lipoprotein assembly. It ties membrane physics (leaflet imbalance and curvature) to specific molecular players (TMEM41B, CLCC1, APOE) and to physiologically important outcomes — VLDL secretion and liver health. Given prior links between TMEM41B/APOE and diverse diseases (viral infection, neurodegeneration, cardiometabolic disorders), identification of CLCC1 as a recruiter/regulator of scrambling opens new mechanistic and therapeutic avenues. Researchers studying lipid metabolism, ER biology, lipoprotein assembly and metabolic disease will find immediate relevance; clinicians and translational scientists may see future biomarker or target potential.
Author take
Punchy: this is a big one. The authors have found a previously underappreciated ER regulator — CLCC1 — that helps localise and boost scramblase activity where the bilayer is most stressed. The study directly links membrane leaflet imbalance to failed APOB lipidation, loss of circulating lipoproteins and rapid liver disease. If you care about how membranes shape metabolism (and many of us should), the detailed data here are worth digging into.
Why should I read this?
Short answer: because it explains why some lipid droplets go rogue and how cells normally stop that from wrecking lipoprotein secretion and the liver. It’s written for people who want a mechanistic bridge between membrane biophysics and metabolic disease — and it points to a new protein (CLCC1) you’ll probably see in follow-up studies. Also, the imaging + proteomics + in vitro reconstitution is satisfying if you like neat, multi-angle evidence.