Homologous recombination deficiency and hemizygosity drive resistance in breast cancer
Article Date: 04 March 2026
Article URL: https://www.nature.com/articles/s41586-026-10197-0
Article Image: Figure 1
{kind=link}
Summary
This large clinicogenomic study (MSK cohort: 6,927 tumours from 5,881 patients) links germline homologous recombination deficiency (HRD), especially gBRCA2, and pre-existing allele-specific copy-number states to predictable routes of acquired resistance in breast cancer. Key findings: gBRCA2 tumours are more likely to harbour RB1 loss-of-heterozygosity (LOH) before treatment, acquire RB1 loss-of-function (LoF) under CDK4/6 inhibitor (CDK4/6i) selective pressure, and progress faster on CDK4/6i plus endocrine therapy (ET). HRD promotes the specific mutational patterns (short indels and structural variants with microhomology) that generate RB1 second hits. Preclinical patient-derived xenografts (PDXs) validate that RB1 loss drives CDK4/6i resistance in gBRCA2 tumours while PARP inhibitors (PARPi) remain effective.
Key Points
- gBRCA2 carriers (and HRD tumours) show enrichment for pre-treatment RB1 LOH and acquire RB1 LoF after CDK4/6i exposure.
- Patients with HR+/HER2– metastatic breast cancer and gBRCA2 had significantly shorter PFS on CDK4/6i + ET (median ≈ 9–12 months vs 15–18 months in gWT cohorts).
- Acquired RB1 LoF in gBRCA2 tumours often bears HRD-associated signatures (short deletions flanked by microhomology), implicating microhomology-mediated end joining.
- Pre-treatment RB1 haemizygosity (single functional RB1 allele) strongly predisposes tumours to acquire a second RB1 hit under CDK4/6i — lowering the evolutionary barrier to resistance.
- PARP inhibitors given after CDK4/6i produced meaningful responses in many gBRCA2 cases; the data suggest PARPi might be preferable earlier in the sequence for this subgroup.
- PDX models from BRCA2 carriers reproduce clinical findings: CDK4/6i resistance associates with RB1 loss, while PARPi sensitivity is retained.
Content summary
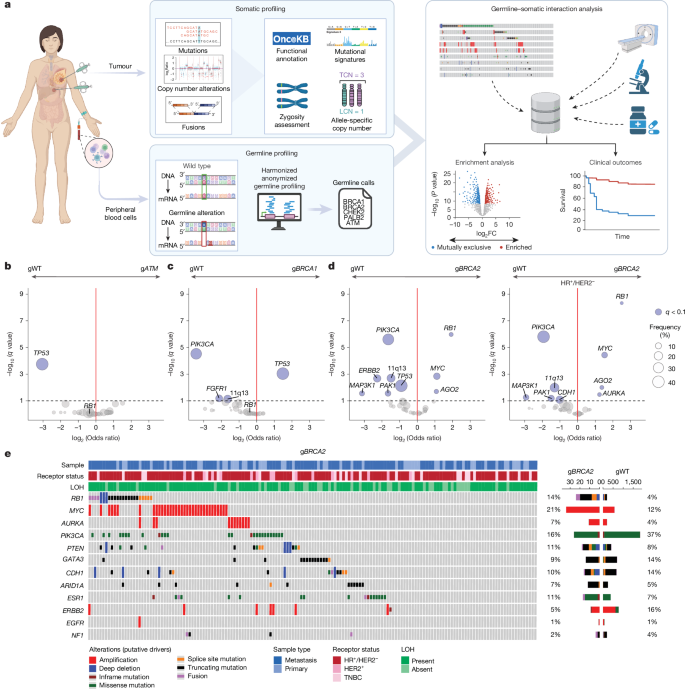
The authors integrated matched germline and somatic sequencing with detailed clinical annotations to explore how inherited variants shape evolutionary routes to therapy resistance. They focused on canonical HRD-associated genes (BRCA1/2, PALB2, ATM, CHEK2) and used allele-specific copy number (ASCN) analyses to infer zygosity and LOH.
Major analyses included germline–somatic interaction testing, survival analyses (PFS/OS) for patients treated with CDK4/6i + ET, a paired pre-/post-CDK4/6i cohort to track acquired alterations, HRD signature inference, and validation in independent real-world datasets and PDX models.
Findings: gBRCA1/2 and other germline PVs shape somatic landscapes (for example, TP53 enrichment in gBRCA1). Specifically, gBRCA2 tumours were enriched for somatic RB1 alterations, MYC and AURKA amplifications — alterations associated with CDK4/6i resistance. In survival models, gBRCA2 status predicted shorter PFS on CDK4/6i + ET; this was replicated in an external multi-institutional cohort. In matched samples, acquired RB1 LoF was significantly more frequent in gBRCA2 tumours after CDK4/6i exposure. The mechanism appears twofold: (1) co-deletion of BRCA2 and RB1 (both on chr13q) producing pre-existing RB1 haemizygosity, and (2) ongoing HRD mutagenesis that generates RB1 second hits, often as short indels or large SV deletions. PDX experiments confirmed that RB1 loss confers resistance to CDK4/6i while PARP inhibition remains effective.
Context and relevance
This study reframes treatment sequencing for a molecularly defined subgroup. CDK4/6 inhibitors are standard first-line therapy for HR+/HER2– advanced breast cancer, but in tumours with BRCA2-driven HRD (or other HRD signatures) the evolutionary trajectory under CDK4/6i is biased toward rapid emergence of RB1 loss and consequent resistance. The result is clinically shorter PFS on standard CDK4/6i + ET. Conversely, PARPi targeted at HRD produce robust responses and may prevent the HRD-mediated path to RB1 loss if given earlier. The work supports using pre-treatment ASCN (for example RB1 haemizygosity) and HRD signatures to stratify patients and design early interception or alternative sequencing strategies (a hypothesis being tested in the EvoPAR-Breast01 trial).
Why should I read this?
Short version — because it changes how you might think about sequencing therapies. If a tumour has BRCA2-driven HRD or RB1 haemizygosity, the usual CDK4/6-first approach risks fast failure. The paper gives you concrete genomic markers (RB1 LOH/haemizygosity, HRD signatures) that forecast a predictable resistance route and points toward upfront PARP strategies for those patients. It’s jam-packed with clinically actionable signals and validated in models — handy if you want to avoid being blindsided by resistance.
Author style (punchy)
Clear, data-rich and clinically provocative. The authors combine massive clinicogenomic datasets with matched pre/post samples and PDX validation to make a tight mechanistic case: pre-treatment allelic state plus HRD biology can predict not just whether resistance will emerge, but exactly how. If you care about precision sequencing of breast cancer therapies, this is must-read evidence that genomics should guide initial treatment choice, not only later salvage decisions.