
A sorghum pangenome reference improves global crop trait discovery
Article metadata
Article Date: 11 March 2026
Article URL: https://www.nature.com/articles/s41586-026-10229-9
Article image: Figure 1
{kind=link}
Summary
This paper presents a 33-member sorghum pangenome anchored on a greatly improved BTx623 V5 reference assembly and high-coverage re-sequencing of nearly 2,000 genotypes. Using long-read assemblies, deep RNA-seq annotation and a pangenome graph approach, the authors catalogue large structural variants, presence–absence and copy-number variation, and link these to phenotypes and geographic/climatic origin. They demonstrate practical pangenome-enabled genotyping (an 80-mer k-mer method) for complex loci such as SHATTERING1 (SH1) and the dhurrin biosynthetic gene cluster (BGC), showing how structural and haplotype variation maps to domestication history, drought adaptation and defence-related chemistry.
The work pairs genomic resources with standardised trait data (field and controlled environments), and spatial analyses to show how migration, human-mediated gene flow and drought prevalence shape allele sharing. The pangenome and k-mer genotyping are pitched as accessible tools to accelerate trait discovery and transfer of useful alleles into breeding programmes, especially in decentralised, climate-adaptive networks.
Key Points
- BTx623 V5 is a major upgrade: 10 chromosomes represented by 34 contigs, far fewer gaps and corrected misassemblies versus V3, improving mapping of key adaptive genes.
- The 33-member pangenome nearly doubles sequence space versus the single reference and reveals hundreds of large structural variants and multiple >1 Mb inversions relevant to trait variation.
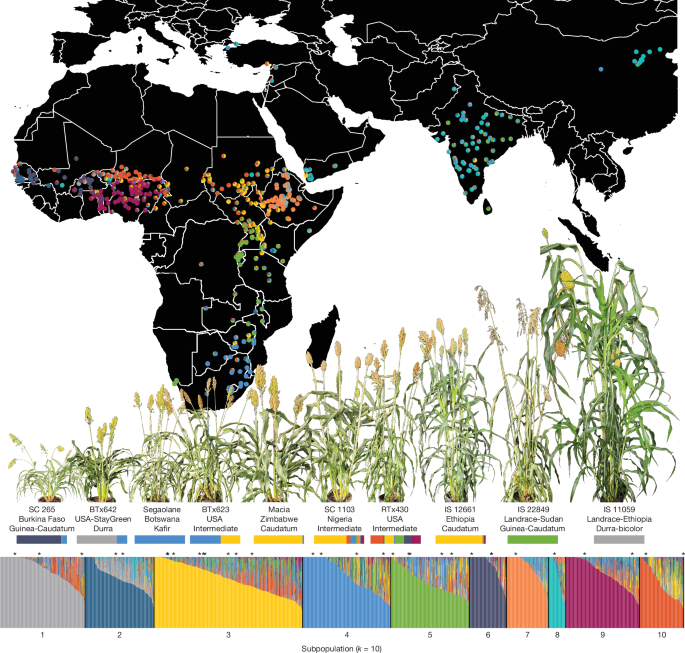
- A diversity re-sequencing panel of 1,984 unique genotypes captures broad phenotypic variation (biomass, flowering, WUE) and ten genetic subpopulations across global germplasm.
- k-mer (80-mer) genotyping provides a computationally light and robust way to type complex SV haplotypes across short-read datasets without rebuilding full graph alignments each time.
- Case studies: SH1 haplotypes show deep divergence consistent with multiple domestication events and clear botanical/climatic structure; the dhurrin BGC contains linked structural and SNP variation that correlates with dhurrin levels and aridity.
- Landscape genetics show long-distance, human-mediated allele sharing, and drought prevalence increases allele sharing of drought-adaptive variants across continental scales.
- Resources (assemblies, annotations, pangenome graph, VCFs) are publicly available via Phytozome / EMBL-ENA and SRA for broad community use.
Content summary
The authors produced long-read assemblies and deep RNA-seq annotation for 33 representative sorghum genotypes, including model, elite and locally adapted lines. They improved the canonical BTx623 reference (V5) and constructed chromosome-level pangenome graphs to capture sequence diversity that short-read mapping misses.
They paired these references with a large short-read re-sequencing panel (1,984 unique genotypes) and standardised trait measures (field biomass, flowering, UAV indices, greenhouse WUE, dhurrin and HCN potential). Through combined pangenome and population analyses they identified structural and gene content variation affecting domestication loci, stress response and secondary metabolism.
Practically, they developed an 80-mer k-mer genotyping pipeline to score diagnostic haplotype sequences in short-read libraries. This was used to classify SH1 haplotypes across the panel, revealing geographic structure tied to botanical types, and to cluster dhurrin BGC haplotypes that predict metabolite levels and associate with drier collection localities.
Context and relevance
This work addresses a major bottleneck in applying genomics to crop breeding: a single linear reference misses many large, breed-relevant variants. By providing a pangenome plus trait-linked reference genotypes and a lightweight genotyping approach, the study gives breeders and geneticists tools to detect functional alleles that matter for local adaptation — drought tolerance, pest defence and flowering time — and to move those alleles into improvement programmes without losing locally valued traits.
It is especially relevant to those working on climate-resilient breeding, pre-breeding for smallholder contexts, or anyone using genomic selection and GWAS in crops with deep population structure. The publicly available assemblies and k-mer methods also lower the barrier for labs that lack heavy graph-genotyping compute resources.
Author style
Punchy: this is a community-scale resource paper with clear, practical outputs — improved reference assembly, a multi-genome pangenome, trait-linked reference phenomes and a usable k-mer genotyping workflow. If you work in crop genomics or breeding, the methods and datasets here will be directly useful; if not, it still sets a new standard for how to connect structural-genomic diversity to adaptive traits.
Why should I read this?
Short version: if you care about getting the right alleles into crops for real-world climates (especially drought-prone regions), this paper saves you the headache of blaming missing variants on a single reference. It gives you ready-made assemblies, trait links and a neat k-mer trick so you can spot complex haplotypes in old short-read datasets without rebuilding the universe each time. Handy, practical and relevant.