Targeting FSP1 triggers ferroptosis in lung cancer
Summary
This Nature study demonstrates that disabling the ferroptosis suppressor FSP1 (AIFM2) is sufficient to trigger lipid-peroxide-driven cell death and dramatically restrict lung tumour progression in multiple in vivo models. Using autochthonous KRAS-driven lung adenocarcinoma GEMMs, orthotopic and subcutaneous transplants, and patient-derived xenografts, the authors show that genetic knockout or pharmacological inhibition of FSP1 increases oxidised PUFA-phospholipids, lowers the reduced:oxidised coenzyme Q ratio and induces ferroptosis that can be rescued by lipid radical-trapping antioxidants (RTAs), dietary vitamin E or loss of ACSL4. GPX4 loss also restricts tumour growth, but FSP1 is uniquely upregulated during LUAD progression and is a more tractable therapeutic target with a clearer safety window than GPX4. The FSP1 inhibitor icFSP1 reduced tumour growth and extended survival in pre-clinical models, supporting FSP1 as a promising therapeutic target.
Key Points
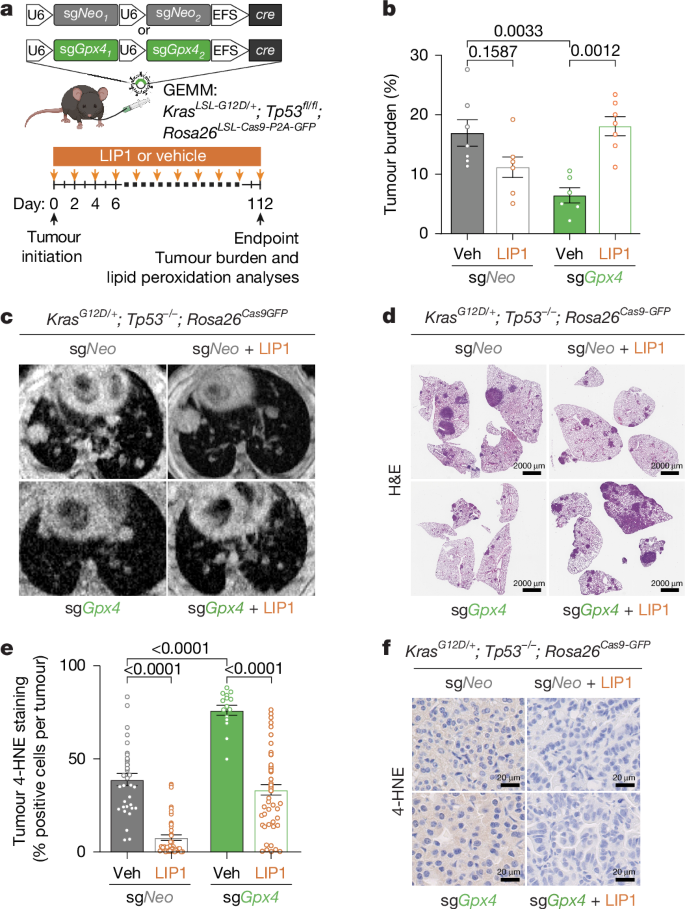
- Genetic deletion of Fsp1 or Gpx4 in KRAS-driven lung adenocarcinoma GEMMs markedly restricts tumour progression by inducing ferroptosis.
- FSP1 expression is upregulated in human LUAD, correlates with higher stage and poorer survival, and increases during tumour progression in mouse models.
- Fsp1 knockout tumours show elevated oxidised PUFA-containing phospholipids and a decreased reduced:oxidised CoQ ratio, consistent with ferroptosis.
- FSP1 dependency is observed across multiple human lung cancer lines and in a pancreatic model in vivo — the effect is cell-intrinsic and not limited to specific oncogenic drivers.
- Ferroptosis suppression (LIP1, vitamin E dietary supplementation or Acsl4 loss) rescues Fsp1 KO tumour growth, confirming lipid peroxidation as the mechanism.
- Pharmacological inhibition with icFSP1 slows tumour growth and extends survival; rescue experiments with an icFSP1-resistant FSP1 mutant show on-target activity.
- GPX4 is essential for tumour cells to avoid ferroptosis, but GPX4 inhibition has toxicity/selectivity issues — targeting FSP1 may offer a better therapeutic window.
Content summary
The authors used CRISPR–Cas9 to knockout Gpx4 or Fsp1 in KP (KrasG12D; Trp53-null) autochthonous LUAD models and observed strong suppression of tumour burden for both knockouts. Lipid peroxidation markers (4-HNE) and targeted oxidised-lipidomics confirmed increased PUFA-PL oxidation in knockout tumours. In vitro, Fsp1 loss did not impair viability unless GPX4 was inhibited, but in vivo FSP1 was essential — Fsp1 KO cells formed much smaller tumours in syngeneic, orthotopic and xenograft models across multiple cell lines and genetic backgrounds. Mechanistic studies showed decreased CoQH2/CoQ in Fsp1 KO tumours and dependence on Fsp1 membrane localisation for protection. Rescue experiments (Acsl4 deletion, dietary vitamin E, LIP1) restored tumour growth, validating ferroptosis as the cause. Finally, icFSP1 treatment phenocopied genetic Fsp1 loss by suppressing tumour growth and extending survival, including in a KRAS-mutant PDX model; effects were blocked by co-treatment with LIP1 or by expressing an icFSP1-resistant FSP1 mutant, supporting an on-target tumour-cell effect.
Context and relevance
Ferroptosis — lethal lipid peroxidation of PUFA-containing membrane phospholipids — has emerged as an exploitable vulnerability in therapy-resistant cancer states. GPX4 and FSP1 are two main defence arms; GPX4 inhibition has shown promise but carries major toxicity and delivery problems. This study is important because it demonstrates, using rigorous in vivo models, that FSP1 is not merely a backup to GPX4 but an independently required protector for lung tumours in the physiological tumour environment. The data suggest a broader, clinically relevant opportunity to target the CoQ–FSP1 axis in LUAD (and possibly other cancers) with fewer side-effect liabilities than direct GPX4 targeting. The translational advance is supported by a tool compound (icFSP1) that has in vivo efficacy and by evidence that FSP1 expression correlates with poor outcome in human LUAD cohorts.
Why should I read this?
Short and blunt: if you care about new ways to kill lung cancers — especially KRAS-driven or therapy-resistant tumours — this paper is a must-see. It shows a clear in vivo Achilles’ heel (FSP1) you can drug, explains the mechanism, proves rescue controls and even tests a clinical-ish inhibitor. Saves you the slog of reading dozens of scattered ferroptosis papers — the authors did the heavy lifting and lay out why FSP1 could be the next drug target to watch.
Author style (punchy)
Strong, rigorous and translational. The work pairs mechanistic lipidomics with robust GEMMs and PDX models, and it delivers a persuasive case that FSP1 inhibition induces tumour-selective ferroptosis and improves survival — a result with immediate therapeutic implications. Read the full paper for the experimental detail and pre-clinical evidence if you’re tracking ferroptosis-targeted therapies.