Lymph node environment drives FSP1 targetability in metastasizing melanoma
Summary
This Nature study reveals that the lymph node (LN) microenvironment reconfigures ferroptosis-defence systems in metastasising melanoma, shifting cells away from GPX4/GSH dependence and toward reliance on FSP1. Using serial LN-selected mouse melanoma lines, RNA-seq, metabolomics, oxygen modulation and in vivo intranodal (i.n.) tumour models, the authors show that low oxygen in LNs promotes GPX4 loss via ubiquitin–proteasome degradation while GCLC expression and glutathione (GSH) levels fall. In contrast FSP1 is upregulated and relocalises to perinuclear lysosomes (via N-myristoylation), creating a context in which pharmacological or genetic inhibition of FSP1 reduces LN tumour growth in vivo. The paper highlights FSP1 inhibitors (viFSP1, FSEN1) as effective at shrinking LN tumours, and suggests therapeutic opportunities for targeting ferroptosis-surveillance switches in metastatic niches.
Key Points
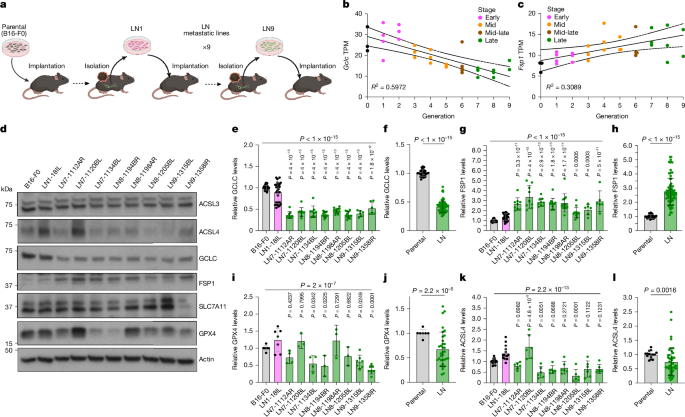
- Serial in vivo LN selection produced melanoma lines with decreased GCLC, GPX4 and ACSL4 proteins and increased FSP1.
- LN microenvironmental hypoxia (≈1% O2) reduces cytoplasmic GPX4 by increasing ubiquitin–proteasome-mediated degradation; GPX4 reduction is reversible on reoxygenation.
- GSH synthesis is impaired in LN-derived lines (lower glutamate, GSH and GSSG), correlating with reduced GCLC expression and epigenetic changes at the Gclc locus.
- FSP1 is upregulated and accumulates at perinuclear lysosomes in LN lines through N-myristoylation; lysosomal FSP1 protects against lysosomal lipid oxidation under ferroptotic stress.
- FSP1 inhibition (viFSP1, FSEN1) as monotherapy reduced intranodal tumour growth in vivo; effects were LN-context dependent and largely on-target (no effect in Fsp1-KO tumours).
- Combining FSP1 inhibition with GCLC inhibition (L-BSO) sensitises LN-derived cells in vitro, though combination benefits in vivo were variable—highlighting context-dependence and pharmacokinetic factors.
- The LN niche creates a durable shift from GPX4 to FSP1 dependence, exposing a metastasis-specific vulnerability that can be therapeutically exploited.
Content summary
The authors generated a panel of LN-metastatic melanoma lines by serially passaging spontaneous LN metastases in mice. Late-generation LN lines show progressive upregulation of Fsp1 and downregulation of Gclc (and reduced GPX4 protein), confirmed by RNA-seq, qPCR and immunoblotting.
Unbiased metabolomics revealed reduced metabolites in the GSH synthesis pathway (glutamate, GSH, GSSG) in LN lines. ATAC–seq indicated reduced chromatin accessibility at the Gclc promoter, and NRF2 levels were lower in late LN lines; NRF2 overexpression raised GCLC, GPX4 and FSP1 levels, suggesting partial transcriptional control of Gclc via NRF2.
Hypoxia experiments showed that low oxygen (1% O2) selectively reduces cytoplasmic GPX4 protein by increasing ubiquitination and proteasomal degradation (rescued by proteasome inhibitors). Paradoxically, low O2 reduces sensitivity to GPX4 inhibitors in cell culture, consistent with LN-mediated protection mechanisms (for example oleic-acid incorporation) and the complex interplay of microenvironmental factors.
FSP1 localisation was characterised: in LN lines FSP1 concentrates at perinuclear lysosomes in an N-myristoylation-dependent manner. Lysosomal FSP1 remains functional across pH changes and limits lysosomal lipid peroxidation; Fsp1-KO cells were more sensitive to lysosome-directed lipid-oxidising agents (fentomycins).
Functional studies: Fsp1 genetic knockout or pharmacological inhibitors sensitised LN-derived cells in vitro and, crucially, daily local administration of cross-species viFSP1 or human-specific FSEN1 reduced intranodal tumour burden in mouse models and extended survival in NSG mice. The anti-tumour effects were specific to LN tumours and were absent (or much weaker) in subcutaneous tumours. In spontaneous metastasis assays, Fsp1 deletion decreased the incidence of tumour-draining LN metastasis.
Context and relevance
Ferroptosis is emerging as an actionable route to kill therapy-resistant cancer cells. This paper shows that the LN microenvironment actively rewires ferroptosis surveillance: hypoxia lowers GPX4, and reduced GSH biosynthesis weakens the GPX4 axis, making FSP1 the dominant defence. That creates a metastasis-specific dependency that can be exploited with FSP1 inhibitors. The findings are directly relevant to researchers developing ferroptosis-based cancer therapies, and highlight the need to test candidate drugs in physiologically relevant microenvironments (for example intranodal models) rather than relying solely on standard in vitro assays.
Why should I read this?
Because if you care about stopping metastasis, the take-home is simple: melanoma in lymph nodes flips its antioxidant backup from GPX4 to FSP1 — and that flip is druggable. The paper does the heavy lifting (genetics, metabolomics, localisation and in vivo proof) so you don’t have to read twenty separate studies to get the idea. If you’re into ferroptosis, metastasis biology or translational drug work, it’s a tidy, actionable story.
Author style / Takeaway
Punchy: this is a clear, mechanistic demonstration that microenvironment matters — big time. The LN niche produces a durable switch in ferroptosis defence that makes FSP1 a viable target in situ. The in vivo efficacy of distinct FSP1 inhibitors (viFSP1, FSEN1) and the genetic KO data make this a high-priority lead for follow-up preclinical work. Caveats remain (why in vivo > in vitro efficacy, and combination rules), but the translational potential is strong.