Specificity, length and luck drive gene rankings in association studies
Summary
This Nature paper compares gene prioritisation from genome-wide association studies (GWAS) and loss-of-function (LoF) burden tests across 209 quantitative traits in the UK Biobank. Using population-genetic models and extensive empirical analyses, the authors show that both study designs favour trait specificity but at different levels: burden tests rank genes (and therefore favour trait-specific, long genes), whereas GWAS rank variants (so they capture context-specific non-coding variants and pleiotropic genes). The study also highlights non-biological influences — gene length increases burden-test power, while genetic drift and allele frequency variation shape GWAS hits — and explains why neither method alone reliably ranks genes by true trait importance.
Key Points
- Burden tests (aggregating rare LoF variants) and GWAS prioritise largely different genes despite some overlap.
- LoF burden tests preferentially detect trait-specific genes because selection keeps high-impact (pleiotropic) LoFs very rare; power scales with mutation target size (μL), so longer genes are more likely to be detected.
- GWAS highlight trait-specific variants, which can be non-coding and context specific (tissue, cell type or developmental window), so they often implicate pleiotropic genes via regulatory variants.
- Random genetic drift and resulting allele-frequency differences make GWAS rankings noisy: high-frequency variants are more likely to appear as top hits and can seem pleiotropic for statistical reasons.
- Neither GWAS nor burden-test P-value rankings reliably equal a gene’s true trait importance because of selection-driven flattening and frequency-dependent noise.
- Aggregating signals across variant types (for example AMM or other gene-level heritability methods) better recovers genes with large true effects and helps estimate trait importance.
Content summary
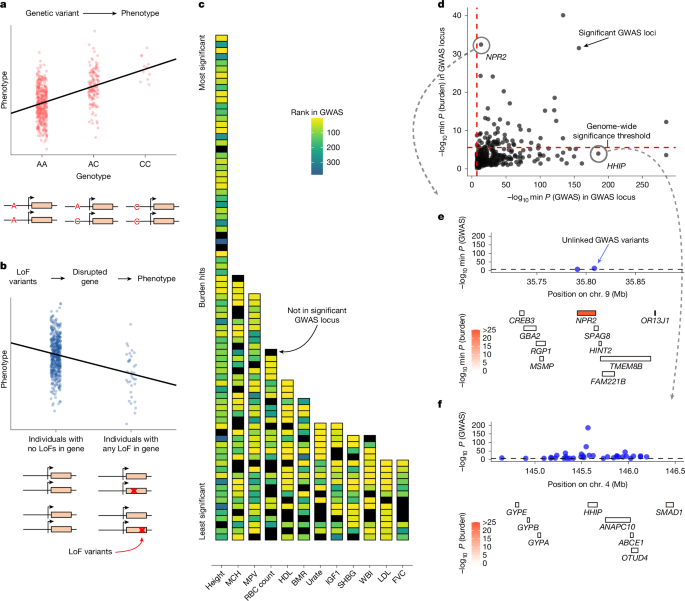
The authors compared GWAS and LoF burden-test results across 209 UK Biobank traits, carefully aligning loci and using conservative locus definitions. They confirmed that although many burden hits fall within GWAS loci, the two approaches rank loci very differently: only about a quarter of genes with burden support fall among top GWAS loci. Using population-genetic theory and simulations, they show that the expected association strength for burden tests scales with gene trait-specificity multiplied by the aggregate frequency of LoF alleles, and because selection keeps high-impact (pleiotropic) genes strongly constrained, burden tests end up prioritising trait-specific genes rather than the largest-effect genes.
For GWAS, the expected signal at a variant is driven by its trait specificity (variant-level), which can arise either because the variant acts through a trait-specific gene or because the variant is context specific (active in the relevant tissue/time). Non-coding, context-specific variants therefore allow GWAS to pick out pleiotropic genes in trait-relevant contexts. The authors used S-LDSC analyses on coding and non-coding annotations and found clear enrichment of heritability in tissue-specific ATAC peaks and in coding variants from specifically expressed genes.
They further demonstrate two non-biological drivers: (1) gene length (μL) boosts LoF burden-test power because more potential LoF sites increase carrier frequency and reduce standard errors; (2) genetic drift causes wide allele-frequency variation, so realized GWAS rankings depend substantially on chance frequency differences. The paper also discusses the flattening effect from selection: very important genes become constrained, making P-value ranking a poor proxy for trait importance. Aggregating signals across variant types (AMM and similar methods) recovers gene-level heritability that better correlates with measures of constraint and importance.
Context and relevance
Why this matters: researchers and drug developers routinely use GWAS or exome burden evidence to prioritise genes. This study clarifies what each approach actually emphasises. Burden-test hits are more likely to be trait-specific and are thus attractive drug targets for fewer side effects, whereas GWAS hits may reveal regulatory mechanisms acting on pleiotropic genes in a context-specific manner. The results argue for combined strategies: use burden tests for trait-specific, coding-impact discoveries and GWAS (plus aggregation methods) to capture regulatory context and total gene-mediated heritability.
Broader relevance: the findings touch on ongoing trends in human genetics — the expansion of large exome and whole-genome cohorts, the emphasis on cell-type-specific regulatory maps, and the push for methods that integrate rare and common variation. The work warns against over-interpreting P-value ranks as measures of biological importance and highlights the utility of Bayesian and aggregation frameworks to improve gene prioritisation for functional follow-up and therapeutic development.
Author style
Punchy — the paper is tightly argued and methodologically careful. If you work on gene prioritisation, translational genetics or mapping regulatory mechanisms, the details are worth digging into: the models explain a lot of paradoxes folks see when comparing GWAS to exome results.
Why should I read this?
Short version: Read it if you pick genes from GWAS or exomes. The paper tells you why those two routes point at different genes, what part is biology (specificity) and what part is artefact (length, luck), and gives practical guidance — aggregated, context-aware methods beat raw P-value rankings when you want the genes that really matter. It’s a tidy, useful reality-check that will save you time and likely change how you prioritise targets.