Adenosine signalling drives antidepressant actions of ketamine and ECT
Summary
This Nature paper shows that rapid antidepressant treatments — a single subanaesthetic dose of ketamine and electroconvulsive therapy (ECT) — produce fast, local surges of extracellular adenosine in mood-regulatory circuits, notably the medial prefrontal cortex (mPFC) and hippocampus. Using genetically encoded GRAB Ado sensors and fibre photometry, the authors demonstrate that these adenosine surges are necessary and sufficient for antidepressant-like effects: genetic or pharmacological loss of adenosine A1 or A2A receptors abolishes efficacy, while activation or local adenosine release in the mPFC reproduces therapeutic effects. Mechanistically, ketamine shifts cellular metabolism (reducing intracellular ATP/ADP and altering mitochondrial TCA flux), driving ENT1/2-mediated adenosine efflux rather than extracellular ATP hydrolysis. The team used adenosine dynamics as a biomarker to screen ketamine derivatives and identified deschloroketamine (DCK) and 2C-DCK as more potent adenosine releasers with antidepressant efficacy at lower doses and reduced hyperlocomotion. They also show that acute intermittent hypoxia (aIH) raises brain adenosine and produces adenosine-dependent antidepressant effects. The work positions adenosine signalling in the mPFC as a unifying, druggable mechanism underlying rapid antidepressant responses and suggests both new compounds and non-pharmacological approaches for major depressive disorder (MDD).
Key Points
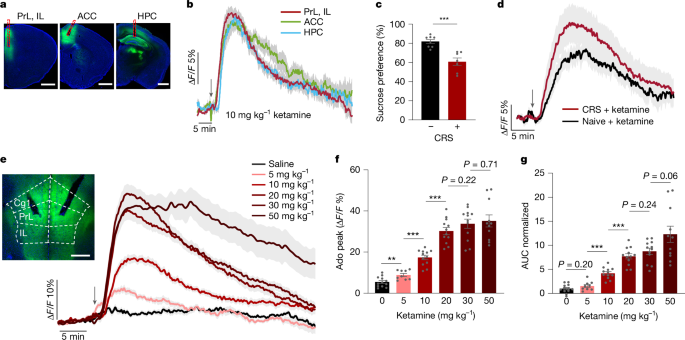
- Ketamine (subanaesthetic dose) and ECT cause rapid, spatially localised surges of extracellular adenosine in mPFC and hippocampus, tracked in real time with GRAB Ado sensors.
- Both adenosine A1 and A2A receptors are required for ketamine and ECT antidepressant effects; receptor activation can mimic treatment outcomes.
- Ketamine-induced adenosine arises from intracellular metabolic changes (reduced ATP/ADP and suppressed mitochondrial pyruvate usage) and is exported via equilibrative nucleoside transporters (ENT1/2), not from extracellular ATP hydrolysis.
- Using adenosine release as a screening biomarker, the authors discovered ketamine derivatives (DCK, 2C-DCK) that evoke stronger adenosine surges, show antidepressant efficacy at lower doses and reduce dissociative-like hyperlocomotion.
- An acute intermittent hypoxia (aIH) protocol elevates brain adenosine and produces adenosine-dependent antidepressant effects without motor deficits—pointing to a noninvasive therapeutic avenue.
- The mPFC is the critical circuit node: local adenosine infusion or astrocyte-driven release in mPFC produces antidepressant-like effects, while mPFC-specific knockout of A1/A2A receptors blocks systemic ketamine benefits.
- Clinical implications include possible interference from caffeine (adenosine antagonism), and opportunities to develop adenosine-centric drugs or non-pharmacological protocols for treatment-resistant depression.
Content summary
The authors combined in vivo optical sensors, genetics, pharmacology, metabolic assays and behavioural tests in mouse depression models. GRAB Ado1.0 recordings show ketamine triggers a dose-dependent adenosine rise in mPFC and hippocampus (but not nucleus accumbens), with kinetics distinct from hypoxia. A1- and A2A-receptor knockout mice failed to show ketamine or ECT antidepressant responses; pharmacological antagonists produced the same loss of effect. Optogenetic activation of astrocytes to produce local adenosine in the mPFC relieved depressive-like behaviours, dependent on CD73. Metabolic probes (PercevalHR) and isolated mitochondrial flux analyses indicate ketamine lowers ATP/ADP and suppresses TCA cycle flux, predicting intracellular adenosine accumulation and ENT-mediated efflux—confirmed because ENT inhibition reduced ketamine-triggered adenosine release. Screening 31 ketamine analogues by in vivo adenosine response identified DCK and 2C-DCK as leads with greater potency and better side-effect profile. ECT also provoked adenosine surges; aIH reliably elevated adenosine and produced receptor-dependent antidepressant effects. Overall, the evidence supports adenosine signalling in the mPFC as a causal, targetable mediator of rapid antidepressant actions.
Context and relevance
This study reframes how rapid-acting antidepressants work: rather than a single receptor target, a metabolically driven adenosine surge (engaging high-affinity A1/A2A receptors in the mPFC) mediates therapeutic action for ketamine and ECT. That matters because it offers a unifying, testable mechanism to guide safer drug design and noninvasive therapies. Using an optical sensor as a functional biomarker to triage ketamine derivatives is a pragmatic translational strategy—it decouples antidepressant efficacy from NMDAR antagonism and may reduce psychotomimetic liabilities. The aIH findings also point to repurposable, non-pharmacological approaches that raise adenosine without seizures. Clinically, factors that block adenosine signalling (for example, caffeine) could blunt these rapid treatments, and adenosine-focused interventions may be particularly relevant for treatment-resistant MDD.
Why should I read this?
Short answer: because this paper probably changes the playbook. Fancy sensors + clever pharmacology show a single neuromodulator — adenosine — ties together ketamine, ECT and even intermittent hypoxia. If you care about rapid antidepressants, mechanism-guided drug design or noninvasive therapies, the data here save you time: it points to actionable targets (A1/A2A in the mPFC), a biomarker you can use for screening, and potential clinical caveats (think caffeine). Read it for the experiments, skim for the translational takeaways.