Whole-genome landscapes of 1,364 breast cancers
Summary
This Nature study reports whole-genome sequencing (WGS) and linked clinical data for 1,364 breast cancers from the Korean CUBRICS cohort. The authors integrated WGS with transcriptomes for most cases and clinical records to map somatic single-nucleotide variants, indels, structural variants and copy-number alterations, and to extract mutational signatures and timing of events.
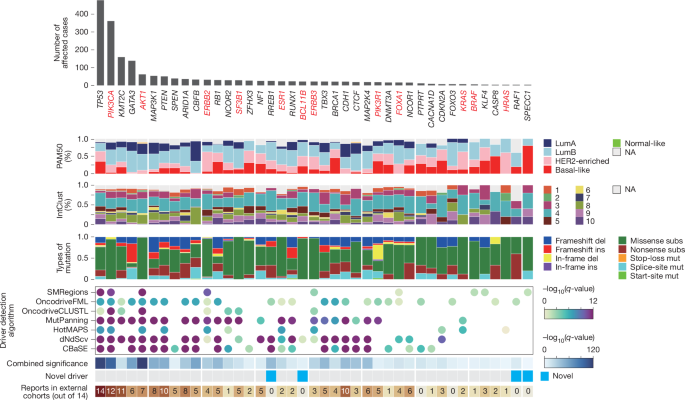
Key findings include identification of 41 driver genes (four putatively novel), widespread homologous recombination deficiency (HRD) signatures in 23% of tumours, frequent structural rearrangements (including enhancer hijacking near ERBB2), recurrent focal amplifications often on extrachromosomal DNA (ecDNA), and numerous recurrent gene fusions (notably more frequent NRG1 fusions than previously reported). The study also links WGS-derived features (MATH heterogeneity score, HRD, TMB, CNA timing) to treatment responses and outcomes, for example HRD predicting better response to adjuvant anthracycline–cyclophosphamide in TNBC but worse progression-free survival with first-line CDK4/6 inhibitor therapy in advanced hormone receptor-positive disease.
Key Points
- Large WGS cohort (n=1,364) with matched clinical records and transcriptomes (n=1,209), median age 44 — distinct Asian population profile.
- 10.9 million somatic mutations identified: SNVs, indels and >200k structural variants; basal-like tumours had highest tumour mutational burden (TMB).
- 41 driver genes discovered (4 novel candidates such as BCL11B and RREB1); TP53 mutations associate with genomic instability and higher intratumour heterogeneity (MATH).
- Comprehensive mutational-signature analysis found 17 SNV, 9 indel and 6 structural-variant signatures; HRD signatures (SBS3, ID6, SV3, SV5) were frequent and serve as WGS biomarkers for PARP inhibitor sensitivity.
- 315 cases (23.1%) classified as HRD; causal homologous-recombination pathway variants found in ~41% of HRD cases (germline BRCA1/2 and somatic RAD51B rearrangements).
- APOBEC3A/3B germline deletion common in this cohort (31.8%), linked to higher TMB and APOBEC mutational signatures but not clearly cancer-predisposing in Koreans.
- Structural variation is pervasive; recurrent translocations bring oncogenes and super-enhancers together (e.g. CCND1–ZNF703/FGFR1 and ERBB2–chr20 loci), suggesting enhancer hijacking drives expression in some cases.
- NRG1 is frequently disrupted by SVs (110 cases) with 17 oncogenic fusions retaining the EGF-like domain — higher frequency than earlier reports.
- Major focal amplifications: ERBB2 (18.7%), CCND1, ZNF703 and FGFR1; >40% of these oncogene amplifications occur on ecDNA; ecDNA presence correlates with F-score and overall tumour evolution.
- ERBB2 absolute copy number (eg ≥33) and transcriptional data improved prediction of pathologic complete response to neoadjuvant anti-HER2 therapy beyond IHC alone; chromothripsis enriched in responders.
- Many long-segment CNAs occur early in molecular time (often before diagnosis), implying decades-long evolution from first genomic events to clinical cancer.
- WGS-derived biomarkers (MATH, TMB, HRD) showed predictive/prognostic associations with PFS and DFS in various treatment contexts (anti-HER2, CDK4/6 inhibitors, adjuvant chemo), warranting prospective validation.
Context and relevance
This work substantially expands WGS-based knowledge of breast cancer by combining genomic breadth with clinical depth in a large Asian cohort. It confirms previously observed processes (TP53-driven instability, APOBEC activity, HRD) while revealing population-specific frequencies (APOBEC3A/B deletion), novel/rare drivers and a higher incidence of certain fusion events (NRG1). The timing analyses and ecDNA observations deepen understanding of tumour evolution and mechanisms of therapeutic resistance. For researchers and clinicians interested in precision oncology, this dataset offers candidate biomarkers and hypotheses that could inform biomarker development, trial design and mechanistic studies.
Why should I read this?
Short version: if you care about what really shapes breast-cancer genomes and what might help pick treatments, this paper is a goldmine. They did whole-genome sequencing on a huge, clinically annotated cohort, found new drivers, showed how structural changes and ecDNA drive expression, and tied WGS features to treatment outcomes. In plain terms — it saves you time: they read the genomes so you don’t have to.
Author style
Punchy: the study is sizable, methodically rigorous and clinically focused — the team doesn’t just catalogue variants, they connect them to treatment response and evolutionary timing. This is high-impact discovery work that should be read in detail by anyone developing genomic biomarkers, designing trials for PARP, CDK4/6 or HER2-targeted therapies, or researching tumour evolution and resistance.