Cross-regulation of [2Fe–2S] cluster synthesis by ferredoxin-2 and frataxin
Article Date: 10 December 2025
Article URL: https://www.nature.com/articles/s41586-025-09822-1
Image: Figure 1
{kind=link}
Summary
This Nature paper shows that frataxin (FXN) and mitochondrial ferredoxin-2 (FDX2) cross-regulate mitochondrial [2Fe–2S] cluster synthesis by competing for the same binding site on the core ISC assembly complex ((NFS1–ISD11–ACP–ISCU2)2). The authors used a physiologically relevant in vitro reconstitution plus biophysical assays (FIDA, ITC), ARBS persulfide assays, AlphaFold modelling and Drosophila genetics.
The main findings are:
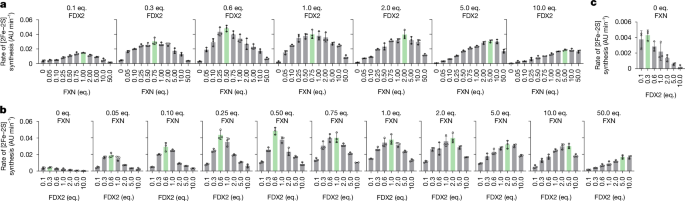
– FXN and FDX2 bind the (NIAU)2 complex with similar affinities and compete in a dose-dependent manner, producing an optimal [2Fe–2S] assembly rate near a ~1:1 ratio.
– Excess FXN slows persulfide reduction by FDX2 and, at very high levels, inhibits overall cluster assembly — explaining FXN overexpression toxicity.
– FDX2 not only competes with FXN but directly inhibits persulfide generation and transfer by interfering with the NFS1 mobile loop; mutational mapping (helix F and C‑terminal residues) identifies residues mediating binding vs repression.
– In a Drosophila Friedreich’s ataxia model, frataxin deficiency upregulates the FDX2 orthologue (Fdx1), and modest knockdown of Fdx1 during development improves fly survival — supporting the idea that reducing FDX2 can partially relieve the assembly defect caused by low FXN.
The authors conclude that mutually exclusive binding and direct inhibitory actions of FDX2 and FXN finely tune Fe–S cluster biosynthesis, and that modulating FDX2 could be explored as a therapeutic axis for Friedreich’s ataxia.
Key Points
- FXN and FDX2 compete for the same binding site on the (NFS1–ISD11–ACP–ISCU2)2 complex; optimal [2Fe–2S] synthesis occurs close to a 1:1 FXN:FDX2 ratio.
- Both proteins show near-equivalent affinities (sub-µM to low-µM range depending on assay), making synthesis highly sensitive to relative protein levels.
- Excess FXN inhibits the FDX2-mediated persulfide reduction step, explaining known toxicity of FXN overexpression in vivo.
- FDX2 also directly represses persulfide generation and transfer by interacting with the mobile loop of NFS1; helix F and C‑terminal residues in FDX2 control binding versus repression.
- In a Drosophila Friedreich’s ataxia model, frataxin deficiency raises Fdx1 expression; partial developmental knockdown of Fdx1 improves lifespan, supporting the in vitro mechanism and suggesting a potential therapeutic angle.
Context and relevance
Fe–S clusters are essential cofactors in many mitochondrial and cellular processes. Friedreich’s ataxia (FA) is caused by FXN deficiency and is clinically severe; current therapeutics focus on restoring FXN but overexpression can be toxic. This work provides a mechanistic explanation for that toxicity (competitive displacement of FDX2 and inhibition of persulfide reduction) and uncovers FDX2 as a modulator that, when tuned, can partially rescue FA phenotypes in vivo. The study uses complementary biochemistry, structural modelling and genetics to bridge mechanism to organismal relevance — relevant for researchers working on mitochondrial biogenesis, Fe–S assembly, and FA drug discovery.
Why should I read this?
Quick answer: if you care about how mitochondria build iron–sulfur clusters or about better ways to treat Friedreich’s ataxia, this paper hands you a tidy mechanism and a surprising therapeutic hint — don’t just boost frataxin blindly. It’s a neat piece of work that pins down why too much FXN can backfire and why dialing back ferredoxin might actually help in certain FA contexts.
Author style
Punchy — the paper is mechanistic and translational. The experimental design links molecular binding and kinetics to structural hotspots and to an animal model, so the results are both convincing and immediately interesting for therapeutic strategy discussions.