Mutations in mitochondrial ferredoxin FDX2 suppress frataxin deficiency
Summary
This Nature study reports that dominant missense mutations in the mitochondrial ferredoxin gene FDX2 (and one mutation in NFS1) partially rescue the growth, development and biochemical defects caused by loss of frataxin in Caenorhabditis elegans. Using a hypoxia-permissive screening approach the authors isolated fdx-2 and nfs-1 alleles that restore iron–sulfur (Fe–S) cluster production enough to boost electron transport chain (ETC) complexes and improve organismal growth. Biochemistry and cell culture work show that excess FDX2 binding to the NFS1-containing Fe–S assembly complex inhibits sulphur transfer; the suppressor mutations weaken that interaction (or reduce FDX2 levels) and thus bypass the need for frataxin to some degree. Importantly, reducing Fdx2 gene dosage (heterozygosity) also ameliorates neurological defects in a frataxin knockdown mouse model, pointing to a potential therapeutic strategy based on restoring stoichiometric balance in the Fe–S assembly machinery.
Author style
Punchy: This is a crisp genetic + biochemical scoop — a classic suppressor screen that reveals a surprisingly simple stoichiometric fix for frataxin loss.
Key Points
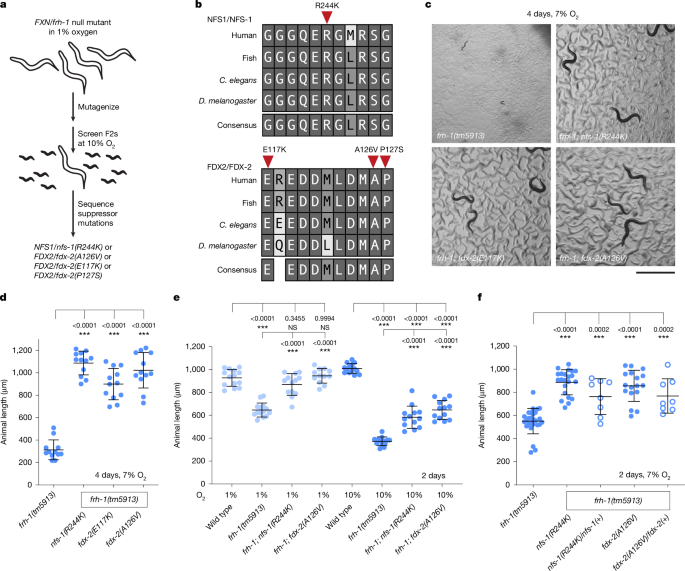
- A forward genetic screen in frataxin-null C. elegans grown in permissive hypoxia identified dominant missense suppressor mutations in fdx-2 (E117K, A126V, P127S) and nfs-1 (R244K).
- Suppressor alleles partially restore Fe–S cluster formation and raise levels of ETC complex I and II subunits, improving growth and development.
- Biochemistry shows FDX2 and frataxin compete for the same NFS1 binding site; excess FDX2 inhibits NFS1 cysteine desulfurase activity and Fe–S assembly in the absence of frataxin.
- Mutant FDX2 variants weaken or alter the FDX2–NFS1 interaction (and in one case can stimulate activity), relieving the inhibition and permitting Fe–S cluster synthesis without frataxin.
- Lowering FDX2 levels (heterozygous null) in worms, and 50% reduction of Fdx2 in a mouse Friedreich’s ataxia model, partially rescues neurological defects — suggesting a therapeutic angle that rebalances protein stoichiometry rather than only replacing frataxin.
Content summary
The authors exploited the fact that frataxin-null C. elegans can survive in hypoxia to perform an EMS mutagenesis screen for rare suppressors that permit growth at higher (non-permissive) oxygen levels. Whole-genome sequencing and CRISPR validation pinpointed specific fdx-2 and nfs-1 point mutations that dominantly improve growth of frataxin-null animals across oxygen tensions.
Proteomics and western blots showed rescue of Fe–S-dependent ETC subunits, especially complex I, and expression of a yeast NADH dehydrogenase (NDI1) confirmed that restoring ETC flux contributes to phenotypic rescue. Structural mapping to cryo-EM models placed the mutations at the NFS1–FDX2 interface. In vitro assays with human proteins revealed that while frataxin activates NFS1 desulfurase activity, excess wild-type FDX2 can displace frataxin and inhibit activity; the FDX2(E131K) variant behaved differently, promoting activity even without frataxin. Overexpression of FDX2 in human cell lines produced Fe–S defects and growth impairment in normoxia that were relieved by hypoxia, mirroring the worm biochemistry.
Finally, the team showed that simple reduction of FDX2 (heterozygous loss) partially rescues frataxin-null worms and improves motor performance in an shFxn mouse model without impacting survival — supporting a model in which adjusting relative levels of FDX2 and frataxin restores Fe–S cluster synthesis.
Context and relevance
Friedreich’s ataxia is caused by reduced frataxin and leads to progressive neurodegeneration and cardiomyopathy. This work reframes part of the problem as a stoichiometric imbalance in the mitochondrial Fe–S assembly complex: when frataxin is low, unopposed FDX2 binding can be inhibitory. That insight shifts therapeutic thinking away from only boosting frataxin levels (which has shown toxicity in some gene-therapy studies) toward strategies that rebalance interacting partners — for example, partial reduction of FDX2 or small molecules that modulate the FDX2–NFS1 interface.
The findings also clarify why hypoxia rescues frataxin deficiency (it alters redox/interaction equilibria or stabilises sensitive intermediates) and connect genetic, structural and biochemical evidence from worms, human cells and mice into a coherent mechanism. For researchers studying Fe–S biology or Friedreich’s ataxia, the paper is directly relevant; for clinicians and translational teams it suggests alternative, potentially safer intervention concepts.
Why should I read this?
Short answer: because the authors found a neat, unexpected hack for a devastating disease. Instead of only trying to replace missing frataxin, they show that easing off the ferredoxin (FDX2) side of the tug-of-war with NFS1 can restore enough Fe–S production to improve function — in worms, cells and mice. It’s a clever mechanism, solidly proven, and it points to fresh therapeutic directions that are worth keeping an eye on.