Insights into DNA repeat expansions among 900,000 biobank participants
Summary
This study analysed short-read whole-genome sequences from about 900,000 participants (490,416 UK Biobank + 414,830 All of Us) to map instability of short tandem repeats (STRs) genome-wide. The authors developed computational methods to extract in-repeat reads (IRRs) and to filter PCR stutter artefacts so they could estimate both germline and somatic repeat-length changes at scale. They focused first on CAG trinucleotide repeats, measured intergenerational mutation rates using identity-by-descent (IBD), and quantified somatic expansions in blood. They then extended the approach to 356,131 polymorphic STRs, identified dozens of unstable loci, performed GWAS for somatic-expansion phenotypes, and linked genetic modifiers—many in DNA-repair pathways—to repeat instability. A notable finding is that highly expanded CAG repeats in the 5′ UTR of GLS associate with liver and kidney biomarkers and elevated risk of renal and hepatic disease, suggesting a dominant, low-penetrance repeat disorder distinct from recessive glutaminase deficiency.
Key Points
- Large-scale analysis: combined short-read WGS from ~900k people (UKB + All of Us) to study repeat instability across the genome.
- Methods: new pipelines to extract in-repeat reads and to filter PCR stutter enabled robust estimates of somatic and germline repeat changes from short reads.
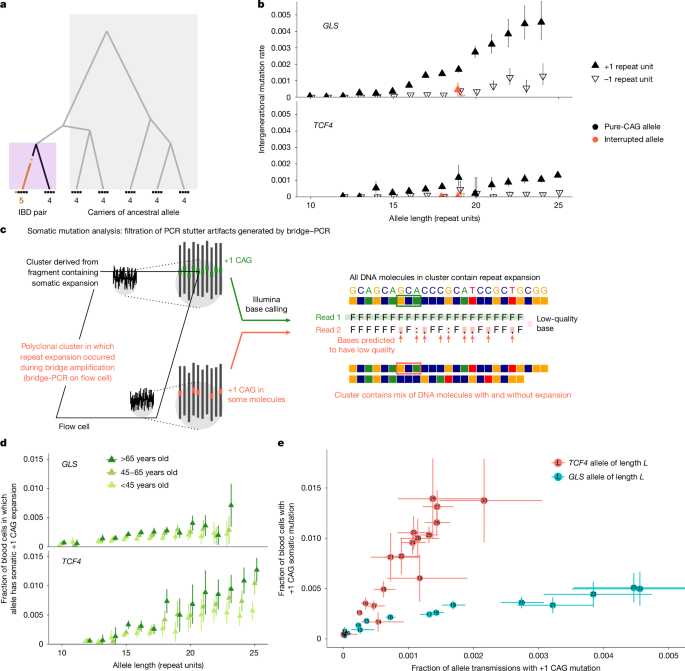
- CAG repeats: most long CAG expansions concentrate at a few loci (notably CA10, TCF4, ATXN8OS); germline mutation rates rise with allele length and are stabilised by repeat interruptions.
- Somatic instability in blood: four CAG loci (TCF4, GLS, DMPK, ATN1) show age-associated somatic expansion; TCF4 is especially unstable in blood and shows extensive mosaicism.
- Genetic modifiers: GWAS and burden tests implicate mismatch-repair and DNA-damage-response genes (MSH3, FAN1, PMS2, MSH2, MLH3 and others) plus XPC, PARP1, NEIL2 and chromatin regulators as modulators of somatic expansion; effects vary across loci and tissues.
- AAAG and other non-CAG repeats: 17 STRs showed clear somatic instability; common AAAG repeats expand with age and are strongly shaped by inherited variation.
- Clinical link: highly expanded GLS 5′-UTR CAG alleles (≈100+ repeats) associate with raised liver and kidney disease biomarkers and much higher odds of severe kidney disease, indicating a distinct pathogenic mechanism (possibly RNA toxicity).
- Implications: somatic instability is under strong genetic control but is locus- and tissue-specific; blood instability can be a biomarker but may not reliably reflect disease-relevant tissues like brain or corneal endothelium.
Content summary
The team began by extracting in-repeat sequencing reads to find long CAG alleles and mapped them to known polymorphic loci. Using IBD sharing they estimated allele-specific germline expansion/contraction rates across 15 well-measured CAG loci, showing higher mutation rates for longer alleles and strong stabilising effects from sequence interruptions. They then devised quality filters to remove PCR stutter from Illumina reads and estimated somatic +1 unit expansion fractions in blood, finding age-dependent increases at TCF4, GLS, DMPK and ATN1. Short-read metrics of long TCF4 alleles (≥45 repeats) were validated with long-read data and used to perform GWAS for somatic expansion in tens of thousands of carriers.
GWAS identified seven genome-wide significant loci for TCF4 blood instability, including MSH3, FAN1, ATAD5 and PMS2; comparisons with modifiers of HTT repeat instability and Huntington’s disease phenotypes revealed both shared and tissue-specific modifier effects, sometimes with opposite directions. Extending their pipeline across 356k STRs, they found 17 loci with evidence of somatic instability (various 2–5 bp motifs); mismatch-repair genes had differing influences depending on motif length (MSH2 stronger on dinucleotides, MSH3 on longer motifs). For AAAG repeats at chr2 and chr19, dozens of common and rare variants (including DNA-repair genes, XPC, PARP1, NEIL2, SMARCAD1) modulate expansion; burden tests also implicated rare pLoF alleles in several repair genes.
Phenome-wide scans linked expansions at multiple loci to diseases and traits. The GLS finding stood out: carriers of very large GLS CAG expansions showed altered liver/kidney biomarkers and markedly higher odds of severe kidney disease and liver disease, replicated across cohorts. The authors propose that highly expanded GLS repeats cause a dominant, low-penetrance disorder via a mechanism distinct from simple loss-of-function (e.g. RNA toxicity), while biallelic GLS LoF causes the known recessive glutaminase deficiency.
Context and relevance
This work leverages unprecedented sample sizes from population biobanks to study STR instability at scale. It confirms that somatic repeat expansion is genetically modulated, implicates many DNA-repair genes as modifiers, and highlights the strong locus- and tissue-specific nature of these effects. For researchers, the findings expand potential therapeutic targets for expansion disorders (beyond HTT) and identify candidate blood-based biomarkers for monitoring somatic instability. Clinically, the GLS association shows how low-penetrance, dominant repeat expansions can be discovered only in very large cohorts and may explain previously unexplained disease risk signals.
Limitations include reliance on short-read WGS (which gives imperfect sizing for very long alleles) and the use of blood as a surrogate tissue; the paper stresses the need for long-read and tissue-specific studies to follow up mechanistic and therapeutic leads.
Why should I read this
Quick take: if you care about repeat-expansion diseases, DNA-repair biology, or biomarker development this paper is big news — it pulls apart how common and rare genetic variation shapes somatic instability across hundreds of thousands of people, and even spots a likely new repeat-driven disease signal at GLS. The tone is punchy: they built clever filters for short-read data, used huge cohorts to boost power, and came away with modifier genes that are plausible drug targets. Read it for the methods, the modifier genetics, and that surprising GLS-clinical link — all of which could shape future translational work.