Critical role for a high-plasticity cell state in lung cancer
Article Date: 21 January 2026
Article URL: https://www.nature.com/articles/s41586-025-025-09985-x
Article image:
Summary
This Nature paper identifies a high-plasticity cancer cell state (HPCS) that emerges early in Kras-driven mouse lung adenocarcinoma and persists through progression. The authors generate faithful reporters (Slc4a11-based) to trace, isolate and ablate HPCS cells in autochthonous models. Functionally, the HPCS acts as a transition hub: it seeds diverse downstream cell states, drives tumour growth and is the principal origin of therapy-resistant minimal residual disease. Ablating the HPCS (genetically or with uPAR-directed CAR T cells) reduces tumour progression and synergises with chemotherapy or KRAS(G12D) inhibition. The HPCS transcriptional signature recurs across multiple human carcinomas and mirrors injury/regeneration epithelial programmes, suggesting broad relevance.
Key Points
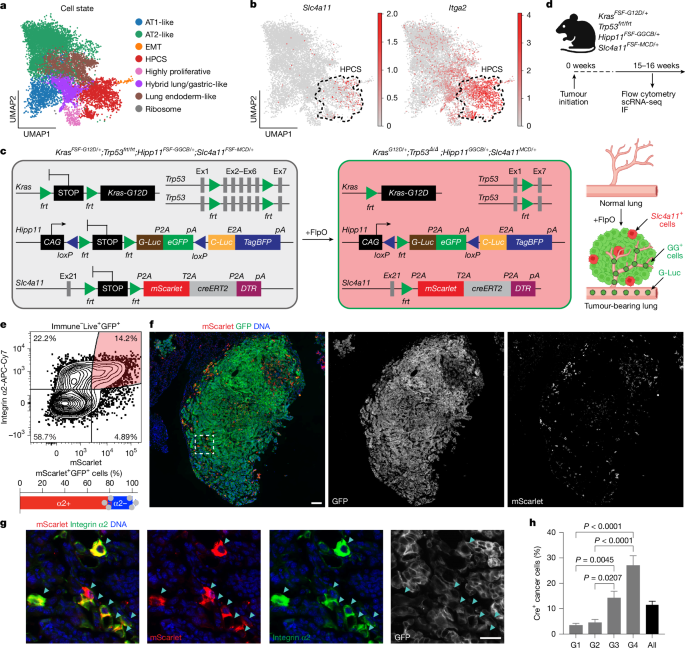
- The HPCS is marked by Slc4a11 and enriched for Plaur (uPAR); a Slc4a11_MCD reporter was developed to trace and ablate it in vivo.
- Lineage tracing shows the HPCS gives rise to all major LUAD cell states and expands phenotypic diversity—it’s a central transition hub rather than a fixed stem cell hierarchy.
- HPCS cells are largely quiescent but produce rapidly proliferating progeny that drive clonal expansion and tumour growth.
- Non-HPCS cells can convert into the HPCS, so the state is dynamically acquired and not a fixed lineage only maintained by self-renewal.
- Ablation of the HPCS impairs progression of early neoplasias, reduces advanced tumour burden and decreases cell-state heterogeneity; combining HPCS ablation with chemotherapy or KRAS inhibition yields near-complete tumour eradication in models.
- Under therapy the HPCS seeds drug-resistant states and MRD; targeting its marker PLAUR (uPAR) with CAR T cells effectively reduces HPCS and tumour growth in preclinical models.
- The HPCS signature correlates with plastic/stress-like programmes across multiple carcinomas and overlaps injury-associated regenerative epithelial states, pointing to a conserved, targetable programme.
Content summary
Using Kras(G12D)/p53-deficient genetically engineered mouse models and sophisticated dual-reporter systems (secreted luciferases plus fluorescent/ablation cassettes), the authors map LUAD evolution at single-cell resolution. They define Slc4a11 as a highly specific HPCS marker and build Slc4a11_MCD and Hipp11_GGCB reporters to trace HPCS cells over time and to selectively ablate them with diphtheria toxin receptor expression.
Lineage-tracing experiments demonstrate that HPCS-labelled cells rapidly exit that state and differentiate into all observed tumour cell states—both alveolar-like and progressed endoderm-like lineages—yielding a marked increase in phenotypic volume. By contrast, lineage-traced differentiated AT1-like cells (Hopx+) show low plasticity and clonal contraction.
Functional tests show HPCS-derived lineages have far higher growth potential than random or AT1-like-derived cells, yet the HPCS itself is mostly quiescent. Importantly, targeted ablation of Slc4a11+ HPCS cells suppresses progression from small neoplastic nodules to larger lesions and reduces markers of aggressive states. In established tumours, HPCS ablation causes regression and reduces heterogeneity.
Therapy studies reveal that, while standard treatments clear many non-HPCS-derived tumour cells, HPCS-derived cells survive and seed therapy-resistant states and MRD. Combining HPCS ablation with cisplatin or KRAS(G12D) inhibitor MRTX1133 eradicates tumours in preclinical allograft models. The authors exploit high Plaur/uPAR expression in the HPCS to test uPAR CAR T cells and show anti-tumour efficacy and HPCS depletion.
Context and relevance
Plasticity is a key cause of therapeutic resistance and intratumour heterogeneity. This paper provides experimental evidence that plasticity concentrates in a specific, targetable cell state—the HPCS—which both seeds diversity and creates therapy-resistant populations. The finding that the HPCS transcriptional programme mirrors injury-associated regenerative states offers a mechanistic explanation (co-option of a transient repair programme) and suggests a therapeutic window since the programme is absent in homeostatic tissue.
Translationally, uPAR (PLAUR) emerges as a viable antigen for cell therapy approaches; combined targeting of the HPCS plus standard therapy may prevent or eliminate MRD. The recurrence of the HPCS-like signature across multiple carcinoma single-cell datasets implies broader applicability beyond LUAD.
Why should I read this?
Short version: if you care about why tumours adapt and resist treatment, this study actually points to a concrete, targetable culprit—the HPCS. It’s packed with clever lineage tools, solid functional data and a translational twist (uPAR CAR T). Read it if you want a clean story linking plasticity, progression and a realistic therapeutic strategy—no fluff.
Author style
Punchy: this is a high-impact, mechanistic and translational study. It’s essential reading for researchers working on cancer plasticity, resistance, single-cell mapping and immunotherapy translation. For clinicians and translational scientists it flags a new tactical target (HPCS/uPAR) for intercepting progression and MRD.