PAF15–PCNA exhaustion governs the strand-specific control of DNA replication
Article metadata
Article Date: 28 January 2026
Article URL: https://www.nature.com/articles/s41586-025-10011-3
Article Image: https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41586-025-10011-3/MediaObjects/41586_2025_10011_Fig1_HTML.png
Summary
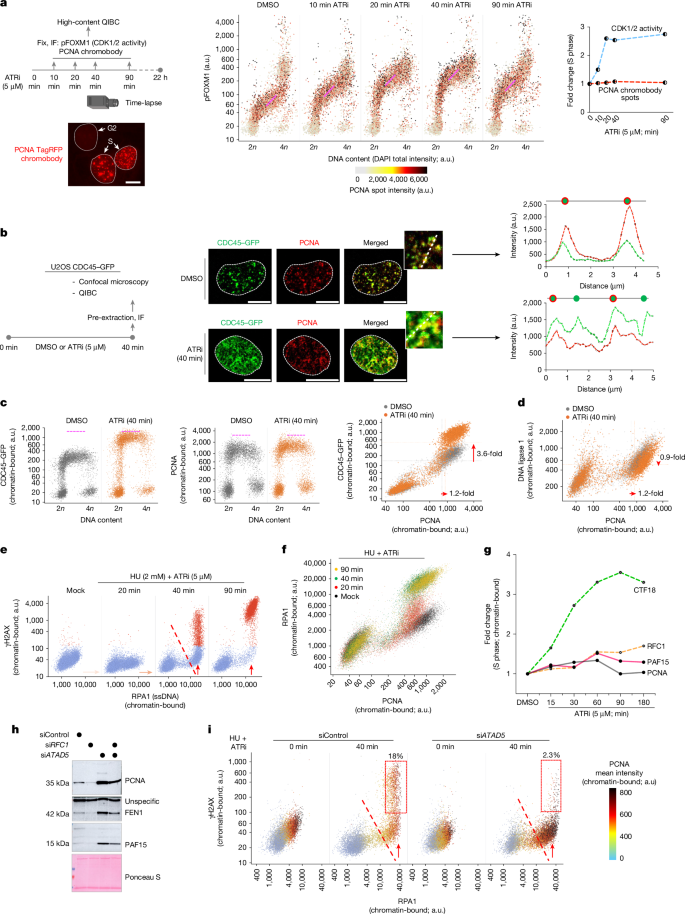
This Nature paper identifies PAF15 as a dosage-limited, lagging-strand-specific factor that stabilises PCNA on chromatin and is essential for canonical Okazaki-fragment (OkF) maturation. The authors show that brief ATR checkpoint inhibition (or other means that drive unscheduled origin firing) causes rapid origin activation that outstrips lagging-strand capacity: chromatin PCNA and PAF15 are functionally exhausted, FEN1 and DNA ligase 1 engagement falls, and PARP1-dependent backup processing is triggered. Loss of PAF15 destabilises PCNA (via premature ATAD5-mediated unloading), causes unligated OkFs, fork asymmetry and replication stress, and sensitises cells to PARP inhibition. Conversely, acute PAF15 excess or mislocalisation (for example to leading-strand PCNA when Timeless/Claspin are lost) is toxic. Transcriptional control by E2F4 keeps PAF15 levels in check; dysregulation links to cancer phenotypes. The work ties replisome component availability to checkpoint control and genome stability.
Key Points
- PAF15 is a low-abundance PCNA partner that preferentially binds PCNA on the lagging strand and helps coordinate Okazaki-fragment maturation.
- Brief ATR inhibition causes rampant origin firing that does not increase chromatin PCNA proportionally, revealing a bottleneck in lagging-strand processing.
- Excess origin firing and PAF15 depletion both lead to reduced chromatin engagement of DNA ligase 1 and FEN1, accumulation of unligated Okazaki fragments and activation of PARP1-dependent repair pathways.
- PAF15 acts as a molecular wedge within the PCNA ring (modelling and structural data) and increases PCNA–DNA affinity, protecting PCNA from ATAD5-mediated unloading.
- Depleting ATAD5 restores PCNA stability and lagging-strand factor engagement in PAF15-deficient cells, demonstrating the unloading mechanism.
- Overexpressing PAF15 or allowing it access to leading-strand PCNA (e.g. after Timeless/Claspin loss) disrupts replisome coordination, slows forks and is lethal — so PAF15 dosage is tightly constrained.
- E2F4-mediated transcriptional repression limits PAF15 levels; loss of E2F4 raises PAF15 and transiently rescues some replication defects but causes long-term instability unless PAF15 is subsequently adjusted.
- Implication: strand-specific replisome capacity — governed by a limited pool of PAF15 — is a rate-limiting control that the ATR checkpoint enforces to avoid catastrophic replication failure.
Context and relevance
Accurate genome duplication requires balanced origin firing and coordinated leading- and lagging-strand synthesis. This study reveals a previously underappreciated, strand-specific limiting factor (PAF15) that determines lagging-strand capacity. Because replication stress and checkpoint failure are central to cancer biology and to the mechanism of several chemotherapeutics, these findings are highly relevant to researchers studying genome stability, replication dynamics, DNA repair and cancer vulnerabilities. The mechanistic link between PAF15, PCNA stability and ATAD5 unloading offers potential axes for therapeutic targeting — for example, exploiting PAF15/PCNA balance in tumours with high origin density or checkpoint defects.
Why should I read this
Short version: because it tells you why suddenly firing lots of origins breaks replication — not just by running out of nucleotides or RPA but by exhausting a strand-specific clamp regulator (PAF15) that keeps the lagging strand working. If you care about replication forks, replication stress, PARP biology or cancer therapy strategies, this paper saves you time by showing the who, what and how — and points to new vulnerabilities.
Author style
Punchy: the authors combine imaging, proteomics, sequencing and structural modelling to make a tight, compelling case that a scarce, strand-specific factor sets a hard limit on genome replication. For anyone working on replisome regulation this is must-read material — the experiments are rigorous and the conclusions shift how we think about checkpoint function and replisome resource allocation.