
Astrocytes enable amygdala neural representations supporting memory
Article metadata
Article Date: 11 February 2026
Article URL: https://www.nature.com/articles/s41586-025-10068-0
Article image: Figure 1
{kind=link}
Summary
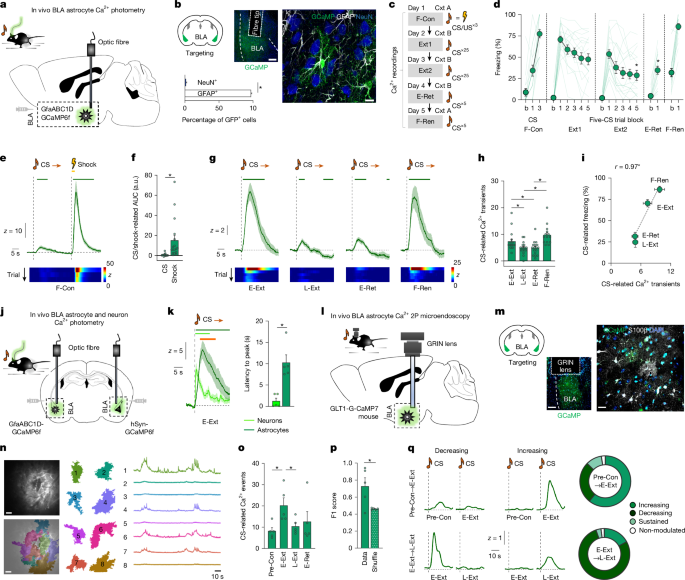
This Nature paper shows that astrocytes in the basolateral amygdala (BLA) actively track and support fear memory retrieval and extinction. Using in vivo calcium imaging (fibre photometry, two-photon and one-photon microendoscopy), chemogenetic and genetic manipulations (hM3Dq, hM4Di, CalEx), optogenetic phototagging and electrophysiology, the authors demonstrate that astrocyte Ca2+ dynamics mirror behavioural fear states and are required for appropriate neuronal encoding of conditioned stimuli. Disrupting astrocyte Ca2+ signalling alters the number and dynamics of CS-responsive BLA neurons, reduces information carried per neuron, impairs readout to prelimbic cortex (PL) projecting neurons and bidirectionally changes fear behaviour and extinction. The work positions astrocytes as causal contributors to the neural representations that underpin memory for threat, not merely passive support cells.
Key Points
- BLA astrocytes show stimulus‑linked Ca2+ signals: strong responses to unconditioned shocks during conditioning and to the conditioned tone during early extinction (fear retrieval), with responses diminishing during extinction and returning on renewal.
- Single-event two-photon imaging reveals heterogeneous astrocyte subpopulations whose CS‑responsiveness can increase, decrease, persist or remain absent across learning and extinction.
- Chronic Ca2+ depletion (CalEx) and temporally precise chemogenetic manipulations (hM3Dq, hM4Di) in astrocytes bidirectionally alter CS‑elicited freezing and disrupt extinction formation, showing causal influence on behaviour.
- Disrupting astrocyte Ca2+ signalling reduces the number of conditionally CS‑excited BLA neurons, lowers ensemble dimensionality and diminishes CS decoding performance — astrocytes enable robust neuronal representations of fear memory.
- Astrocyte manipulations impair encoding and readout specifically in BLA→prelimbic cortex projecting neurons, linking astrocyte activity to cortical readout that drives behavioural expression of fear.
- Findings suggest neuromodulators (for example, noradrenaline) and gliotransmission are likely mediators of astrocyte contributions to memory; targeting astrocytes could offer translational routes for pathological fear.
Author style
Punchy: this is not a marginal tweak to existing models — it forces a rethink. The paper supplies strong causal and population-level evidence that astrocytes do more than support neurons: they shape the neural codes in the amygdala that actually carry fear memory and guide extinction. If you follow circuit-level memory or glia research, the detailed imaging, decoding and projection-specific readout data are worth digging into.
Why should I read this?
Quick answer: because it flips the script on who’s running the show in fear memory. Want to know how non-neuronal cells change what neurons represent about danger — and how that affects behaviour? These experiments stitch together imaging, manipulation and decoding across cells and circuits so you don’t have to. If you work on memory, emotion, glia or therapeutic targets for anxiety disorders, this saves you weeks of digging through piecemeal studies.
Context and relevance
The study sits at the intersection of glia biology and systems neuroscience. It builds on accumulating evidence that astrocytes carry behaviourally relevant signals and extends this to show they are necessary for the fidelity and plasticity of neuronal ensemble codes in the amygdala. The results matter for models of memory that have been neuron‑centric for decades and have implications for strategies to treat pathological fear and PTSD by targeting astrocyte-linked signalling or neuromodulatory routes (for example, noradrenergic pathways).