
CLCC1 promotes hepatic neutral lipid flux and nuclear pore complex assembly
Article Date: 25 February 2026
Article URL: https://www.nature.com/articles/s41586-025-10064-4
Article Image: Figure 1
{kind=link}
Summary
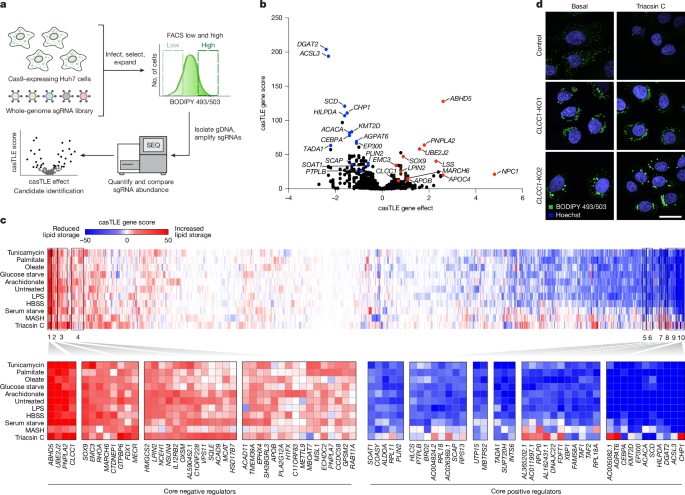
This Nature paper identifies CLCC1 as a key regulator that links nuclear envelope membrane remodelling to hepatocyte neutral lipid handling. Using genome-wide and targeted CRISPR–Cas9 screens in hepatoma cells, the authors flagged CLCC1 as a strong modifier of neutral lipid storage. Loss of CLCC1 causes hepatocytes to accumulate unusually large, PLIN2-negative lumenal lipid droplets (LDs) that are MTP-dependent and fail to be secreted as normal apoB-containing lipoproteins. Liver-specific Clcc1 knockout in mice drives steatosis, reduced plasma TAG and apoB-containing lipoproteins, and liver injury markers.
Structurally, CLCC1 contains a conserved MFH (membrane fusion h-shaped) domain homologous to yeast Brl1/Brr6. CLCC1 forms disulfide-stabilised dimers and higher-order oligomers. Molecular dynamics and mutagenesis indicate a lumenal amphipathic helix (AH2) and oligomerisation are required for CLCC1’s ability to remodel membranes. Loss of CLCC1 also impairs nuclear pore complex (NPC) assembly, producing nuclear envelope herniations and fewer functional NPCs. The authors propose CLCC1 is a metazoan homologue of Brl1/Brr6 that acts as a lumenal membrane fusogen, coupling NPC insertion and neutral lipid flux in hepatocytes.
Key Points
- Genome-wide and metabolic-state CRISPR screens in Huh7 cells identified CLCC1 as a core negative regulator of neutral lipid accumulation.
- CLCC1 knockout hepatocytes accumulate large PLIN2-negative lipid droplets that are trapped in the ER lumen and resistant to cytosolic and ER lipases.
- Liver-specific Clcc1 deletion in mice causes hepatic steatosis, enlarged livers, elevated hepatic TAG and cholesteryl esters, and a near absence of circulating apoB-containing lipoproteins.
- The aberrant lumenal LDs in CLCC1-deficient cells are MTP-dependent and enriched in apoB, but they are far larger than normal VLDL particles and are poorly secreted.
- CLCC1 is structurally homologous to yeast Brl1/Brr6 (the MFH domain) and is required for nuclear pore complex assembly — CLCC1 loss yields nuclear envelope herniations and reduced NPC number and transport.
- Molecular dynamics and mutational analysis show CLCC1 oligomerisation and a lumenal amphipathic helix (AH2) drive membrane bending and fusion-like intermediates; mutations in these regions fail to rescue NPC and LD phenotypes.
- Phenotypes overlap with ER scramblase (TMEM41B/VMP1) and torsinA-related pathways, suggesting interplay between membrane remodellers, scramblases and NPC/lipoprotein biogenesis in hepatocytes.
Context and relevance
This study bridges two fields often studied separately: nuclear envelope/NPC biogenesis and hepatic lipid metabolism. By identifying CLCC1 as a conserved membrane-remodelling factor that both promotes NPC insertion and controls the partitioning of neutral lipids between cytosolic LDs and lumenal lipoproteins, the work suggests a mechanistic link between membrane fusion events in the ER/nuclear envelope and lipid trafficking in hepatocytes. The findings have immediate relevance to understanding hepatic steatosis and defective lipoprotein secretion — processes central to MASLD/MASH and metabolic disease — and point to CLCC1 and its interacting machinery (MTP, scramblases, torsinA) as potential nodes for therapeutic exploration.
Why should I read this
Short version: this paper explains how one membrane protein, CLCC1, pulls double duty — it helps build nuclear pores and decides whether liver cells stash fat or ship it out. If you care about liver disease, lipoprotein secretion or how membrane fusion is controlled in the secretory pathway, this saves you hours of methods-heavy reading and gives you the key discoveries up front.